
Each month, we highlight new research from the scientific community that advances our understanding of complex genetic diseases and showcases the tools researchers rely on for precise variant interpretation. November’s publications explore three distinct phenotypes: congenital cataract, glaucoma, and fetal lymphatic or venous abnormalities, revealing how specific genetic mechanisms shape early development and ocular health. Together, these studies highlight the importance of integrated analysis approaches, including VarSeq, in investigating rare variants and refining genotype–phenotype relationships.
Title: Abnormal Splicing in the Final Intron of PRX Results in Dominant Congenital Cataract Without Neurological Phenotype
Background: Congenital cataract often arises from disrupted lens fiber differentiation, but a causative variant is still only identified in about half of hereditary cases despite >50 known cataract genes. PRX is classically linked to recessive demyelinating neuropathies through loss-of-function variants in L-periaxin (L-PRX), yet L-PRX is also expressed in the lens cortex adherens junction, suggesting additional tissue-specific roles.
Objectives: To determine how heterozygous variants affecting splicing of the final intron of PRX contribute to autosomal dominant congenital cataract without neurological manifestations through clinical, genetic, and functional characterization.
Subjects and Methods: Four unrelated families with unexplained congenital cataract underwent exome (families 1–2) or genome (families 3–4) sequencing. Variants were assessed by segregation analysis, population frequency (gnomAD v4.0), and in-silico prediction tools (CADD, SpliceAI, Pangolin). Candidate PRX splice-region variants were functionally tested using a PRX minigene construct (exon 5–7 of PRXb / exon 6a of PRXa) expressed in HEK293 cells, with RT-PCR and Oxford Nanopore single-molecule sequencing used to define splicing outcomes. Exome data for two families were interpreted using SNP & Variation Suite and VarSeq (Golden Helix, Bozeman, MT, USA).
Results:
- Identified heterozygous splice-region variants in the final intron (intron 6) of PRXb (L-PRX) in seven affected individuals from four families: c.382-2A>C (Brazilian family, de novo and segregating), c.382-3C>A (Portuguese sisters), c.382-1G>A (British proband, de novo), and synonymous c.381G>C (British mother and son). All variants were absent from gnomAD v4.0.0.
- In-silico prediction and minigene assays demonstrated consistent disruption of intron 6 splicing for all alleles, including intron retention and use of alternative donor or acceptor sites. These changes generated a mixture of transcripts: increased PRXa (S-PRX) due to intron retention, and abnormal PRXb (L-PRX) isoforms with in-frame deletions within the nuclear localization signal or C-terminal truncations.
- Clinically, all variant carriers presented with autosomal dominant congenital cataract without reported peripheral neuropathy, despite PRX’s established role in myelination. Literature review identified additional cataract-associated PRX variants clustering in the same splice region, further supporting a specific lens phenotype distinct from previously reported recessive neurological PRX alleles.
- Because loss of L-PRX alone does not cause cataract in humans or mice, the data suggest that excess S-PRX and/or mutant L-PRX acts via gain-of-function or dominant-negative effects on lens cortex adherens junction complexes, rather than simple haploinsufficiency, thereby expanding the phenotypic spectrum associated with PRX.
Conclusions: Variants that disrupt splicing of the final intron of PRX define a novel mechanism for dominant congenital cataract without neurological disease. By shifting PRX isoform balance toward S-PRX and generating aberrant L-PRX proteins, these alleles appear to exert toxic effects on lens development and transparency, distinct from the loss-of-function mechanism underlying recessive neuropathies. Recognition of this PRX intron 6–centered mechanism broadens the genotype–phenotype correlations for PRX and supports consideration of PRX analysis in families with unexplained congenital cataract.
How VarSeq Was Used: “This human study was approved by the Institutional Review Board at Children’s Wisconsin and the Medical College of Wisconsin; and the HRA committee of East of EnglandCambridge South (REC 14/EE/1112) for patients and relatives recruited for the Genomics England 100,000 Genomes Project (100KGP) (familes 3 and 4). The study protocols observed the tenets of the Declaration of Helsinki; written informed consent was obtained from each study participant or their parent. Exome sequencing for families 1 and 2 was performed by Perkin Elmer and analyzed as previously described utilizing family analysis in SNP & Variation Suite and VarSeq software (Golden Helix, Bozeman, MT, USA). Families 3 and 4 underwent genome sequencing as part of the 100KGP, including the clinical variant interpretation pipeline (The National Genomics Research Library v5.1; Genomics England, London, UK) as previously described. Variants were confirmed by Sanger sequencing for families 1 and 2. In silico analysis of variants of interest was performed using gnomAD v4.0.0, CADD 1.6, SpliceAI, and Pangolin.”
Title: WT1–MMP9 regulatory axis and its modulation by TNF-α in early and late onset glaucoma phenotypes
Background: PCG and PACG lie at opposite ends of the glaucoma spectrum but share ECM dysregulation, anterior segment defects, and inflammatory signaling. How transcriptional regulators and cytokines connect these phenotypes is still unclear.
Objectives: To investigate how WT1, MMP9, and TNF-α form a shared regulatory axis linking early-onset PCG and late-onset PACG.
Subjects and Methods: Targeted panel sequencing was performed in 586 PCG patients negative for known candidate genes, 687 relatives, and 1,757 controls. PACG cohorts were evaluated for association of the TNF-α promoter SNP rs1800629. Functional work included wt1/pax6 knockdown and CRISPR disruption in zebrafish, ChIP and dual-luciferase assays in HTM cells, and TNF-α, leptomycin B, L-NAME, and LPS treatments to probe nitric oxide–dependent WT1 nuclear export, MMP9 regulation, and in vivo inflammatory effects.
Results:
- Identified 10 candidate genes in PCG, including 62 rare WT1 variants and 45 rare MMP9 variants; four pathogenic WT1 variants clustered in the nuclear export sequence, and three pathogenic MMP9 variants (one co-occurring with WT1) showed genotype–phenotype correlations.
- wt1 knockdown in zebrafish produced PCG-like ocular defects, pax6 knockdown caused severe iris anomalies, and combined sub-effective wt1/pax6 knockdown reintroduced eye and systemic defects, supporting a genetic interaction in anterior segment development.
- In HTM cells, WT1 and PAX6 bound the MMP9 promoter; WT1 repressed MMP9, while TNF-α induced nitric oxide–dependent nuclear export of WT1, relieving repression and increasing MMP9, with the patient-derived WT1 P264L variant impairing this translocation.
- The TNF-α promoter SNP rs1800629 (A allele) associated with PACG and showed higher promoter activity, and LPS-induced inflammation in zebrafish increased mmp9 and neurodegeneration markers, both rescued by NOS inhibition, indicating a conserved TNF-α–NO–WT1–MMP9 axis.
Conclusions: WT1 acts as a nodal regulator integrating developmental (WT1–PAX6) and inflammatory (TNF-α–NO) inputs to control MMP9 and ECM remodeling across PCG and PACG. This shared WT1–MMP9–TNF-α axis links early and late glaucoma phenotypes and highlights WT1 localization and MMP9 modulation as potential therapeutic targets.
How VarSeq Was Used: “Standard analysis packages of GATK and Varseq were used to identify the potential variants. Initially, the involvements of the observed variants to PCG were ascertained based on their segregation with the disease phenotype, frequencies in the controls, conservation of the wildtype residues, pathogenicity based on REVEL (rare exome variant ensemble learner) scores along with the the revised ACMG guidelines.”
Title: Homozygous Nonsense Variant in GJA4 Associated With Increased Fetal Nuchal Fold Thickness and Abnormal Ductus Venosus Termination
Background: Connexin-37 (GJA4) is essential for lymphatic valve and endothelial function in animal models, but no human congenital phenotype has been linked to biallelic GJA4 loss-of-function. Increased fetal nuchal translucency or nuchal fold thickness often suggests chromosomal or genetic disease, yet many cases remain unexplained after routine testing.
Objectives: To determine whether a novel homozygous GJA4 nonsense variant contributes to a fetal phenotype of increased nuchal fold thickness and abnormal ductus venosus termination.
Subjects and Methods: A pregnant patient underwent ultrasound, CMA, and whole-exome sequencing, with variant interpretation based on gnomAD frequency, ACMG guidelines, and segregation analysis. STRING network analysis was used to assess biological relevance to lymphatic and vascular pathways.
Results:
- Markedly increased nuchal fold thickness (10 mm), skin edema, and infrahepatic ductus venosus termination identified by ultrasound.
- CMA revealed a region of loss of heterozygosity on chromosome 1p including GJA4.
- WES detected a novel homozygous frameshift variant c.97delC (p.Arg33Alafs*98); both parents were heterozygous carriers.
- Variant extremely rare in gnomAD and absent in ClinVar, meeting ACMG PM2 (VUS).
- STRING analysis showed GJA4 interacting with lymphatic-development genes (e.g., ADAMTS3, CCBE1, FLT4) but not with rasopathy-associated genes.
Conclusions: A previously unreported homozygous GJA4 nonsense variant was identified in a fetus with increased nuchal fold thickness and abnormal ductus venosus drainage, suggesting a potential role for connexin-37 loss-of-function in human fetal lymphatic and venous development. Although classified as a VUS, the findings support further investigation of GJA4 in unexplained fetal edema and ductus venosus abnormalities.
How VarSeq Was Used:
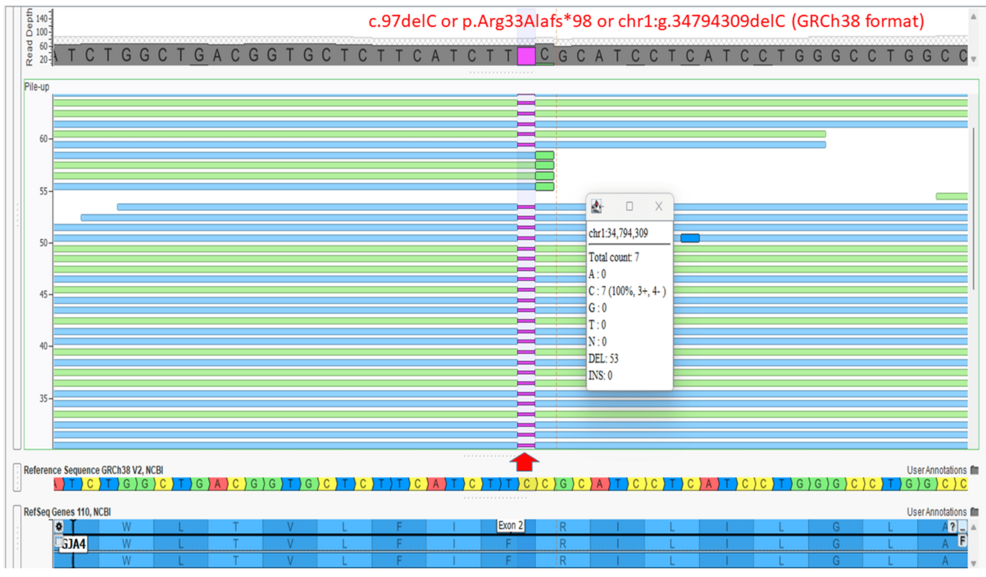
“Figure 4: Screenshot of the Integrated Genomic Viewer after whole-exome sequencing
Screenshot of the Integrated Genomic Viewer (VarSeq; Golden Helix Inc., Bozeman, Montana, United States) of the binary alignment map (BAM) file of the whole-exome sequence data for the affected fetus shows a homozygous variant in the GJA4 gene, c.97delC (transcript ID NM_002060.3) or p.Arg33Alafs*98, or chr1:g.34794309delC (GRCh38 format). The position of the variant is shown by a red arrow.”
Citation
This month’s publications highlight how diverse clinical questions, ranging from congenital cataracts and glaucoma to fetal vascular anomalies, can benefit from precise, structured variant analysis. Whether researchers were interrogating splice disruption in PRX, regulatory signaling in WT1–MMP9, or a novel GJA4 loss-of-function allele, each study illustrates how careful interpretation can clarify mechanisms that standard evaluations may miss.
Across these very different phenotypes, the analytical needs remain consistent: robust filtering, reproducible workflows, and clear evidence frameworks. VarSeq supports this range by giving teams a unified environment to assess variants from exome, genome, or targeted data with confidence and consistency. To learn more about how VarSeq supports clinical and research programs by contacting our team today.



