
Aberrant splicing is a major cause of human disease, with an estimated 15–30% of pathogenic variants either disrupting an existing splice site or introducing a novel one [1]. While variants that alter the canonical AG/GT dinucleotides are straightforward to detect, those that affect the broader splice motif or activate cryptic splice sites are considerably harder to identify. To address this challenge, numerous algorithms have been developed to predict when a variant is likely to alter splicing.
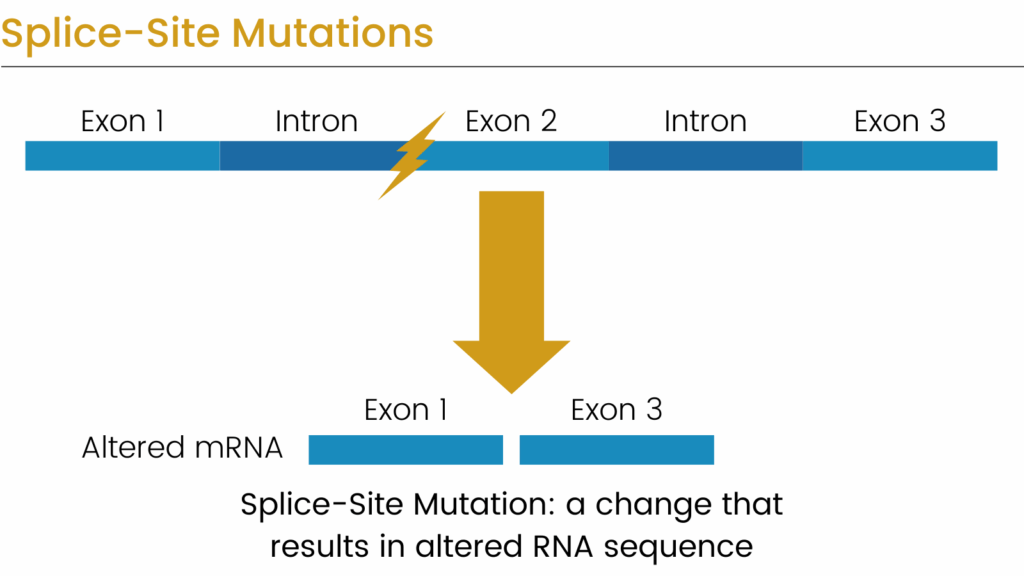
Since its publication in 2019, SpliceAI has become the leading algorithm for identifying splice-altering mutations [2]. However, the restrictive licensing of the original SpliceAI model and its precomputed scores has limited adoption by commercial clinical laboratories. In response, several open-source reimplementations and updated models have been developed.
CI-SpliceAI is one such open-source method that reimplements the SpliceAI algorithm using the more modern TensorFlow library [3]. The CI-SpliceAI authors retrained the model using a collapsed isoform set representative of all manually annotated constitutive and alternative splice sites from GENCODE. The authors compared their method to SpliceAI and demonstrated a small improvement over the original algorithm. We have performed our own independent benchmarking, which demonstrates similar results, showing that CI-SpliceAI is a viable alternative to the original SpliceAI algorithm [4].
These results convinced us to make CI-SpliceAI available to VarSeq users. We are excited to announce the release of new CI-SpliceAI annotation tracks providing precomputed CI-SpliceAI scores for more than 47 million variants computed natively for both GRCh37 and GRCh38 reference assemblies. To ensure compatibility with VarSeq workflows, we updated the transcript annotations used by CI-SpliceAI to match the most recent RefSeq genes annotation tracks. The precomputed tracks include only variants where at least one CI-SpliceAI score exceeds 0.1, and have been enhanced with additional variants from ClinVar and other curated sources to ensure comprehensive coverage of clinically relevant splice-altering variants.
For each variant, CI-SpliceAI provides the following scores along with the relative distance to the novel or disrupted splice site:
- Delta Score Acceptor Gain
- Delta Score Acceptor Loss
- Delta Score Donor Gain
- Delta Score Donor Loss
A Real-World Example
To illustrate the value of CI-SpliceAI relative to the legacy splice-site algorithms currently available in VarSeq, consider the variant NM_000527.5:c.1845+11C>G in LDLR Gene:
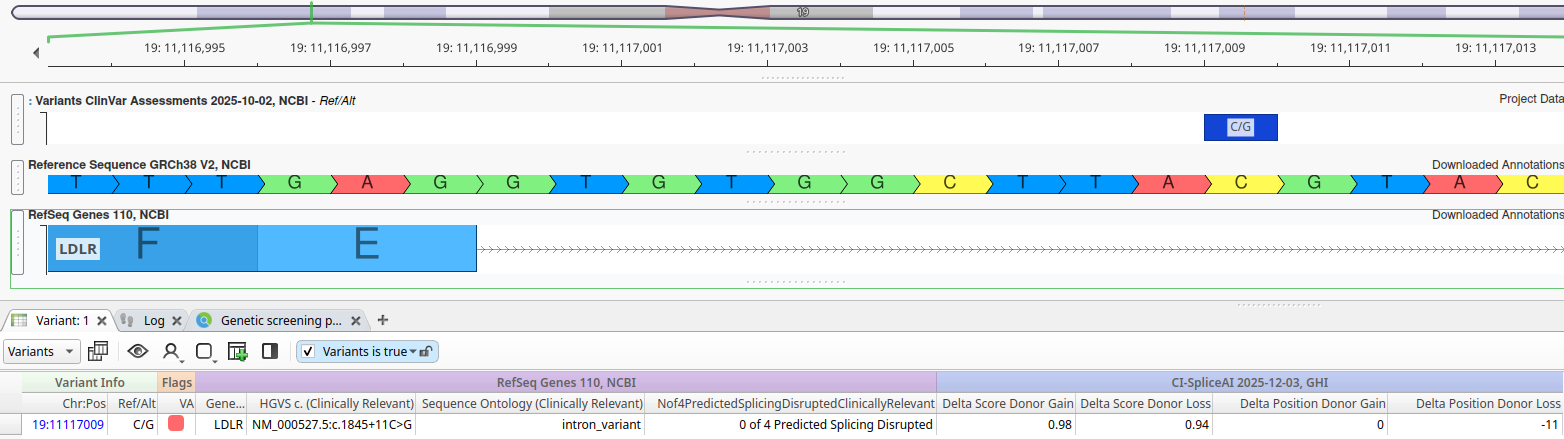
This variant is located 9 base pairs downstream from the canonical splice donor dinucleotide at the exon 12 donor site, and none of the four legacy algorithms predict this splice site to be disrupted. However, the CI-SpliceAI scores paint a different picture:
- Delta Score Donor Gain: 0.98 (Position: 0)
- Delta Score Donor Loss: 0.94 (Position: -11)
CI-SpliceAI predicts with high confidence that this mutation disrupts the existing donor splice site and introduces a cryptic splice site 11 base pairs downstream. Given this new information, let’s examine the reference sequence more closely. The last three base pairs of this newly extended exon would be TAG; thus, if CI-SpliceAI is correct, this variant extends exon 12 and introduces a premature stop codon, which is likely to result in nonsense-mediated decay.
A review of the literature reveals a publication by Graham et al. in which the authors demonstrate that this variant does indeed activate a cryptic splice site in intron 12, introducing a premature termination codon [5]. RNA analysis confirms that this variant results in aberrant splicing and that the resulting mRNA is expected to undergo nonsense-mediated decay. This clearly Pathogenic variant was only detectable through CI-SpliceAI and would have likely been ignored by relying on legacy algorithms alone.
Conclusion
The example above demonstrates how intronic variants that fall outside the canonical splice motifs can have profound functional consequences that traditional algorithms fail to detect. By incorporating CI-SpliceAI scores into your variant analysis workflow, you can identify clinically significant variants that might otherwise be overlooked. With precomputed scores now available for over 47 million variants, VarSeq users can seamlessly integrate this powerful predictive tool into their existing annotation pipelines. To learn more about how you can access the CI-SpliceAI annotation tracks in VarSeq, please reach out to our team.
References
- R. Wang, I. Helbig, A. C. Edmondson, L. Lin and Y. Xing, “Splicing defects in rare diseases: transcriptomics and machine learning strategies towards genetic diagnosis,” Briefings in bioinformatics, vol. 24, no. 5, 2023.
- K. Jaganathan, S. K. Panagiotopoulou, J. F. McRae, S. F. Darbandi, D. Knowles, Y. I. Li, J. A. Kosmicki, J. Arbelaez, W. Cui, G. B. Schwartz and others, “Predicting splicing from primary sequence with deep learning,” Cell, vol. 176, no. 3, pp. 535-548, 2019.
- Y. Strauch, J. Lord, M. Niranjan and D. Baralle, “CI- SpliceAI—improving machine learning predictions of disease causing splicing variants using curated alternative splice site,” PLoS One, vol. 17, no. 6, p. e0269159, 2022.
- N. Fortier, G. Rudy and A. Scherer, “Analyzing the Performance of Deep Learning Splice Prediction Algorithms,” bioRxiv, 2025.
- C. Graham, B. McIlhatton, C. Kirk, E. Beattie, K. Lyttle, P. Hart, R. Neely, I. Young, D. Nicholls, “Genetic screening protocol for familial hypercholesterolemia which includes splicing defects gives an improved mutation detection rate,” Atherosclerosis, vol. 182, no. 2, pp. 331-340, 2005.



