As VarSeq’s adoption has grown among analysts using whole exome data to diagnose rare diseases, a couple of family designs outside of the common trio of an affected child and both parents have come up frequently.
While having both parents provides the maximum power to discover de novo mutations and recessively inherited variants, it is not always possible to contact or enlist parents in the diagnostic odyssey. Thankfully, all is not lost, as a recent review of diagnostic rates of whole exome sequencing showed singleton analyses were still able to achieve a diagnosis rates of 22-25% (compared to 31% for trios). That demonstrates considerable diagnostic utility!
Along with proband-only (singleton) analysis used in the diagnosis of rare recessive disorders, the analysis of large family pedigrees is a common way to tackle rare dominant disorders where multiple affected individuals are filtered against many unaffected, closely related family members to find candidate dominant disease genes.
With VarSeq 1.3.1 we have expanded our algorithms to support these analysis strategies. The new Count Variants by Gene algorithm can support singleton-based compound het searches and Aggregate Counts per Gene enables complex pedigree dominant gene candidate searches.
Gene Level Analysis
Both of these algorithms look to provide some summary of what is going on at a given point in the filtering process (presumably after selecting for rare, potentially functional or damaging mutations) at the gene level for a single sample or a cohort of samples.
What our existing trio-based Compound Heterozygous algorithm is doing is finding genes that contain at least one filtered heterozygous variant inherited from each parent (the “two-hit” nature of recessive disorders).
Without parents, the best one can do in finding these potential autosomal recessive candidate genes is to find the genes that have two or more candidate heterozygous variants.
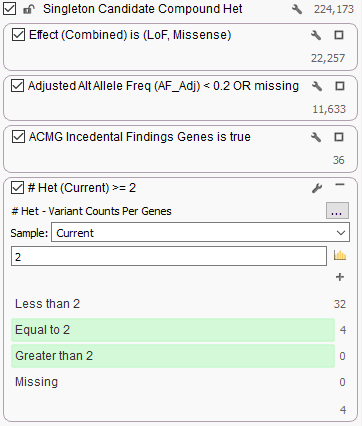
In this example filter chain, we are looking for somewhat rare (<20%) variants in the ACMG Incidental Finding gene list that are potentially gene-altering (Missense or Loss of Function). We count these variants per gene and look only at genes with >= 2 variants.
Running Variant Counts Per Genes on an example exome of a public sample with some not-too strict filter settings, we are able to find a couple genes in the ACMG Incidental Findings gene list that have 2 heterozygous variants for this sample:
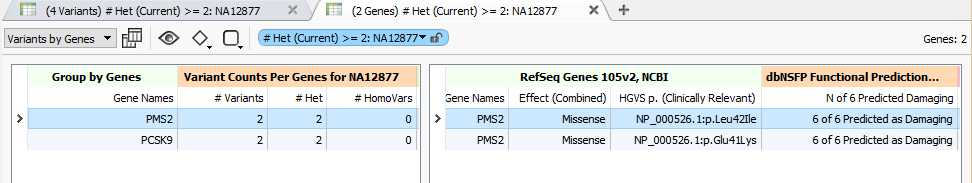
Interestingly, for PMS2 (associated with Lynch syndrome) both of its variants are missense mutations with likely damaging predictions from 6 out of 6 of the functional prediction algorithms in our updated dbNSFP 3.0 annotations.
Although these variants are not in the latest release of the ~60K exomes from the Broad ExAC catalog (which was used as a filter in this project), they are in 6 out of the 17 samples in the CEPH Family 1463 that I was doing this analysis on.
How was I able to easily know that? Well, for one I can visualize these “private” mutations in the built-in GenomeBrowse view in VarSeq:
The 17-member CEPH 1463 family has 6 members with novel mutations in PMS2 (an ACMG Incidental Finding gene linked to Lynch Syndrome). These may be private carrier mutations.
But also, I ran the new Aggregate Counts Per Gene algorithm, which looks at each sample’s filtered variants at a given point in the filtering process, and counts how many samples have variants within a gene as well as how many unique variant sites and total genotypes were observed.
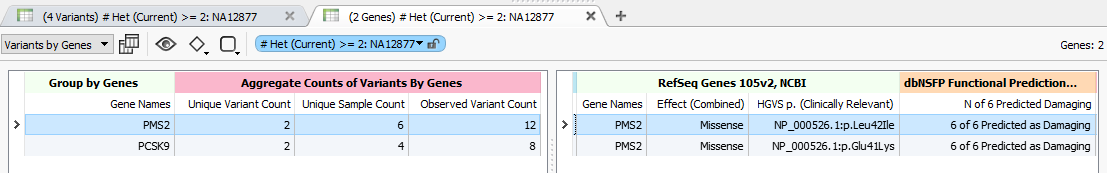
With the aggregate counts, we can see how many samples and observed genotypes have filtered variants for these genes.
Commonly, when searching for dominant model variants, where a single heterozygous variant is linked to a disease, but may have partial penetrance in a family or present in multiple families with different dominant mutations acting on the same gene, it helps to take this gene-centric view across your filtered cohort. In fact, we even allow these counts to be broken out into Affected / Unaffected groupings or other custom sample-wise groupings (maybe family or carrier status based).
With these two algorithms now part of VarSeq, we look forward to seeing a more wide range of workflows being implemented. You may find even more uses than the obvious ones I described here!



