The Golden Helix SNP & Variation Suite (SVS) platform is a powerful and versatile set of tools and algorithms for performing genomic research. That research spans from data originating on genotype micro-arrays to next-generation sequencing. While the majority of SVS users start with genotype data on their samples, any genomic information across a cohort can be used in our various numerical tests including linear and logistic regression. With the latest SVS release, we brought the ability to call Copy Number Variations on targeted gene panel and exome NGS coverage data to SVS and adapted the output for use for association studies and other cohort analysis.
Bringing CNVs to Large-N Workflows
Over the past year, you may have seen the announcements and webcasts about the algorithm we developed in the VarSeq platform for calling CNV events based on the computed coverage over the targets of gene panels and exomes. We have turned and benchmarked this approach against high-quality validated data and recently seen independent validation through some of our clinical customers own benchmarking work. While it serves the needs of clinical labs and other users in the single-sample analysis workflow, we have heard interest in making this product available to the research workflows involving groups of samples being compared or investigated.
To that end, we have added a new premium feature to SVS that enables running the VS-CNV algorithm on all your samples and producing useful output matrices designed for cohort analysis. Fundamentally, the starting point for this algorithm is a BED file defining the regions of your target capture kit (or equivalent imported through our Convert Wizard) and the BAM files for each sample in the cohort. The new SVS-CNV interface can auto-map BAM files to the samples names of your starting spreadsheet or use the spreadsheet itself to define a mapping to BAM file paths.
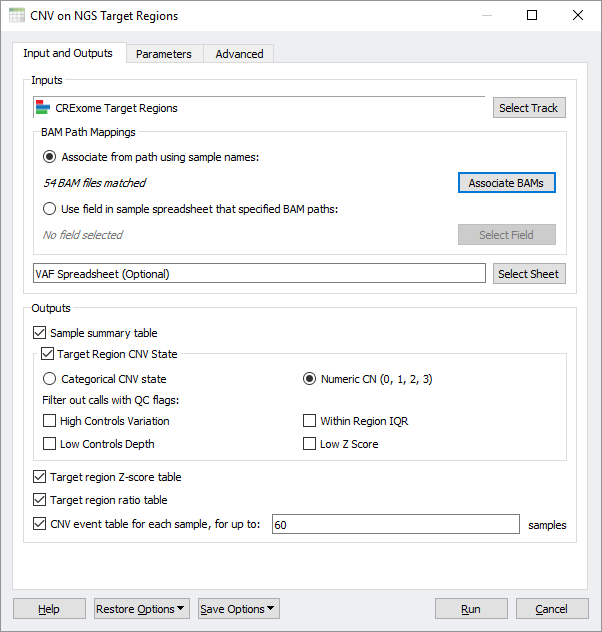
An optional spreadsheet of Variant Allele Frequencies, now easily imported through our VCF import wizard, can be used as an additional supporting data to inform the CNV caller. Most importantly, this dialog allows you to select how you want the algorithm output to be formatted for your downstream analysis. The most significant output is the CNV call state, which encodes numerically by defauly so that it can be used in various analytical methods that scan genomic data. The states here are comparable to numerically encoded genotypes, but in this case, they range from the CNV state of a full deletion to a full duplication (with 2 being the normal state of a diploid region). By having a matrix that is oriented around target regions, we have a uniform filled in matrix based on the units of the targets themselves. The CNV caller algorithm provides some useful QC flags, which can be used to remove lower-quality events and mark their corresponding target states as missing.
We also allow users to export supporting metric data such as the Z-scores and Ratios into matrixes. If you do want to dig into per-sample details of some of your samples, you can generate per-sample CNV event tables, which provide a number of supporting details and metrics of each CNV call, including calculated P-values.
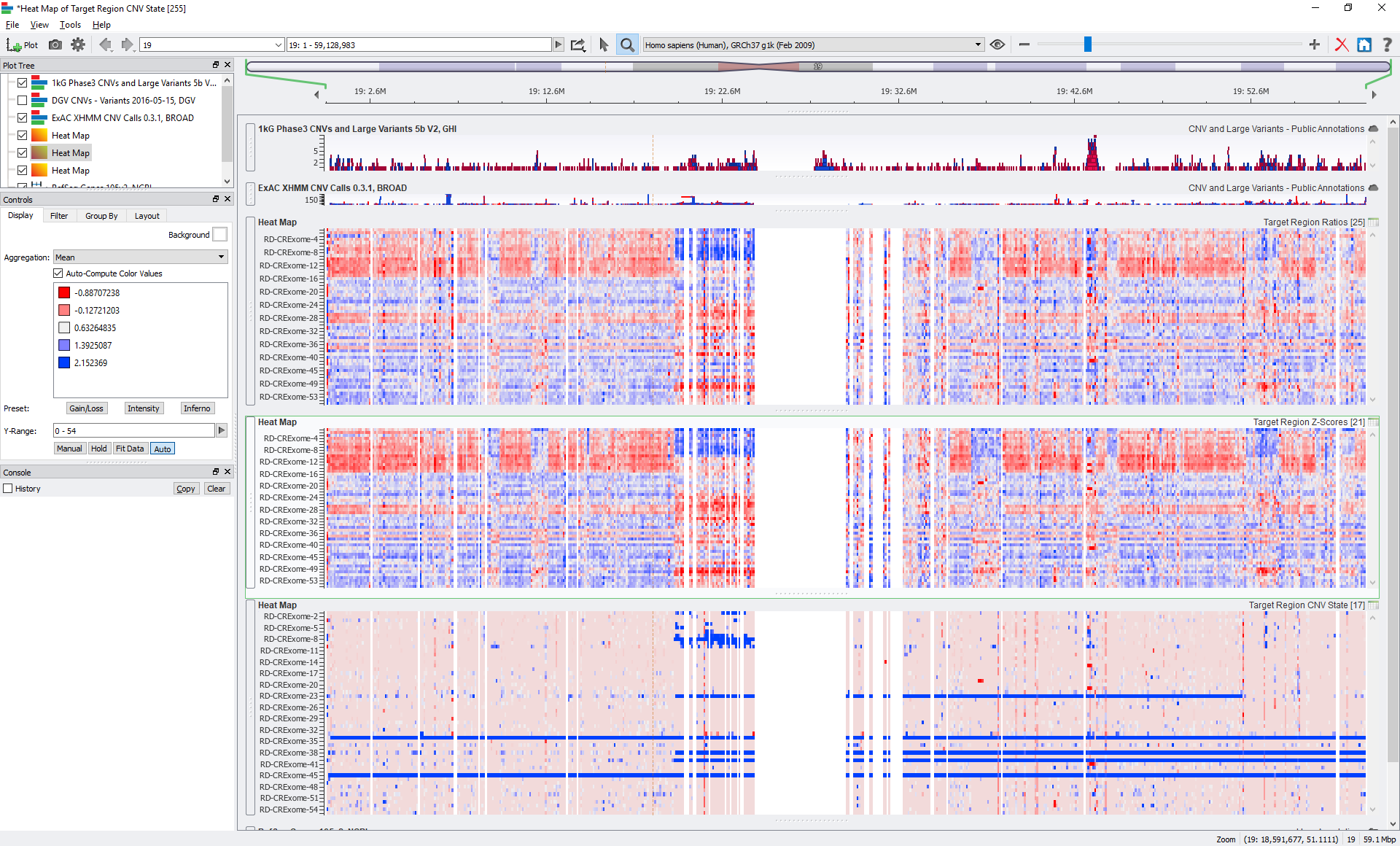
If you would like to see more about this method, please join us in our upcoming webcast or reach out to us to set up a demo by emailing [email protected]!



