This webcast generated some great questions! If you have any other questions for me that are not answered below, please feel free to ask those by emailing [email protected].
Does VSClinical come with support for the new reference genome?
Yes! We worked hard to make everything work in VSClinical regardless of your choice of reference genomes. The only caveat is that CADD annotation, which is not used directly as a recommendation engine component. However, everything else is available on the GRCh38 assembly.
Are there cancer annotations for the new reference genome?
This is a great question – we do have CIViC coming in on a monthly basis that is a fantastic clinical annotation for cancer. The annotations are 37 by default, but we lift over to 38, so we present that on both coordinates. We are also in the process of QC’ing an updated COSMIC! COSMIC is now natively on 38, but we will bring this back to 37. We have a lot of exciting upcoming product ideas and announcements to be had about focusing on the VSClinical-style interpretation of cancer, but we plan to support both 37 and 38 as equal partners. Moreover, as you can see in the webcast, it’s easy to move back and forth to both.
Can you elaborate on the issue of mapping RefSeq transcripts obtained from UCSC? What kind of errors have you encountered?
Note: I got contacted by the bioinformatic folks at the UCSC and informed that now additionally provided RefSeq with NCBI alignments using their “ncbiRefSeq” track (versus the “refGene” table/track that uses BLAT described below).
Essentially, they use BLAT, which is there own fast-alignment algorithm, to place the canonical RefSeq transcript coding sequence onto the genome. It’s not that theirs are necessarily always worse, in most cases that I’ve seen – maybe less than ideal, it’s just that if you’re going to have differences, you might as well go with the NCBI. However, there are examples where the NCBI and UCSC mappings are different, in some cases radically so.
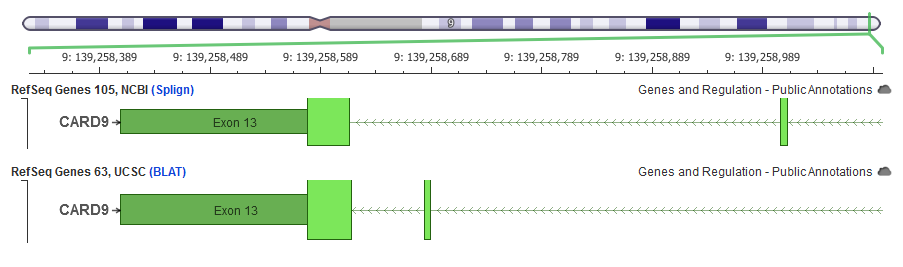
Moreover, then in other cases where I’ve dug in, I’ve generally been happier with the NCBI mappings that we had with this recent release from the interim that they gave us.I did have a previous webcast about transcripts and genome annotations – it’s an old one, but I cover all of this in more detail if you’re interested in checking it out:
I did have a previous webcast about transcripts and genome annotations – it’s an old one, but I cover all of this in more detail if you’re interested in checking it out.
Other than CADD scores, are there other annotations that have yet to be ported to GRCh38?
That’s it! It’s a lot of effort to maintain annotations on both genome assemblies, but as a Golden Helix customer, its part of the package. We have our annotation team set up to use the same tools available to you with our Convert Wizard to move data to whatever genome it is not natively on. So, you get to see the same annotation on both references! When you are in VarSeq, and you go to add annotations, you’re automatically filtered down to the annotations for a given genome assembly, and you should be able to find everything you need there.
Do I have to upload my data to use the LiftOver capabilities?
No, like everything in VarSeq, it runs locally on your computer without any transfer of your data. We have implemented the UCSC LiftOver algorithm strategy right into our core algorithm annotation and importing pipelines, allowing this to seamlessly integrate with your analysis workflow that you control on you local compute!