
Compound heterozygosity is one of the most clinically important, and most frequently missed, mechanisms behind recessive genetic disease. Understanding it is essential for anyone interpreting genomic data, from clinical lab scientists to genetic counselors.
What Is Compound Heterozygosity?
Compound heterozygosity occurs when an individual carries two different pathogenic variants in the same gene, with each variant located on a separate copy (allele) of that gene, one inherited from each parent. Both copies of the gene are functionally disrupted, which is what makes the condition disease-causing.
This is distinct from a classic homozygous recessive scenario, where the same variant appears on both alleles. In compound heterozygosity, the two variants differ; they just happen to reside in the same gene and collectively knock out its function.
Clinically, this distinction matters because compound heterozygous individuals can present with the full recessive disease phenotype even though neither parent is affected.
Compound Heterozygous vs. Heterozygous: What’s the Difference?
The difference between heterozygous and compound heterozygous is subtle but consequential.
A simple heterozygous individual carries one functional allele and one altered allele of a given gene. In most autosomal recessive conditions, a single working copy is enough. The functional allele compensates, and the person is a carrier without symptoms.
A compound heterozygous individual has no fully functional allele. One copy of the gene carries variant A; the other carries variant B. Neither copy works properly. The result is the same loss-of-function outcome as having two copies of the same pathogenic variant, but the route there involved two different mutations.
This is why compound heterozygosity can be so easy to overlook: each parent looks like a healthy carrier on its own. It’s only when you examine both alleles together in the child that the disease mechanism becomes apparent.
A Simple Example of Compound Heterozygosity
Cystic fibrosis provides one of the clearest illustrations. The CFTR gene encodes a chloride channel protein, and loss-of-function variants in both copies of the gene cause disease.
The most common pathogenic variant is F508del, a deletion of three nucleotides that removes a phenylalanine residue at position 508. But CFTR has hundreds of known pathogenic variants, and not every affected individual carries two copies of F508del.
Consider this scenario:
| Allele 1 | Allele 2 | |
| Parent A | F508del | Normal |
| Parent B | Normal | G542X (nonsense variant) |
| Child | F508del | G542X |
Parent A is a carrier for F508del. Parent B is a carrier for G542X. Neither parent has cystic fibrosis. But their child inherits F508del from one parent and G542X from the other, leading to a compound heterozygous genotype that disrupts CFTR function on both alleles and causes cystic fibrosis.
This is compound heterozygosity in practice: two different variants, same gene, opposite alleles, full disease expression.
Compound Heterozygosity Across Variant Classes
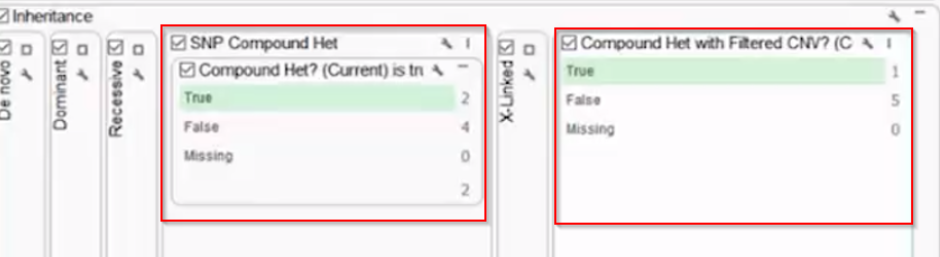
Most people think of compound heterozygosity as a SNP + SNP scenario: two point mutations or small indels, one per allele. That’s the classic case and the easiest to detect. But compound heterozygosity can also occur across different variant classes, and this is where clinical interpretation gets significantly more complex.
SNP + SNP
This is the most straightforward case. Both variants are detected by the same sequencing pipeline, annotated together, and relatively easy to phase. Standard variant filtering and family-based analysis can usually identify these pairs without specialized tooling.
SNP + CNV
This cross-class scenario involves one allele carrying a sequence-level variant (e.g., a missense or nonsense mutation) and the other carrying a copy-number variant, typically a deletion that removes one or more exons. One variant is detected by variant callers, the other by CNV analysis tools. They often appear in entirely separate output files, making it easy for one to be reported while the other is missed.
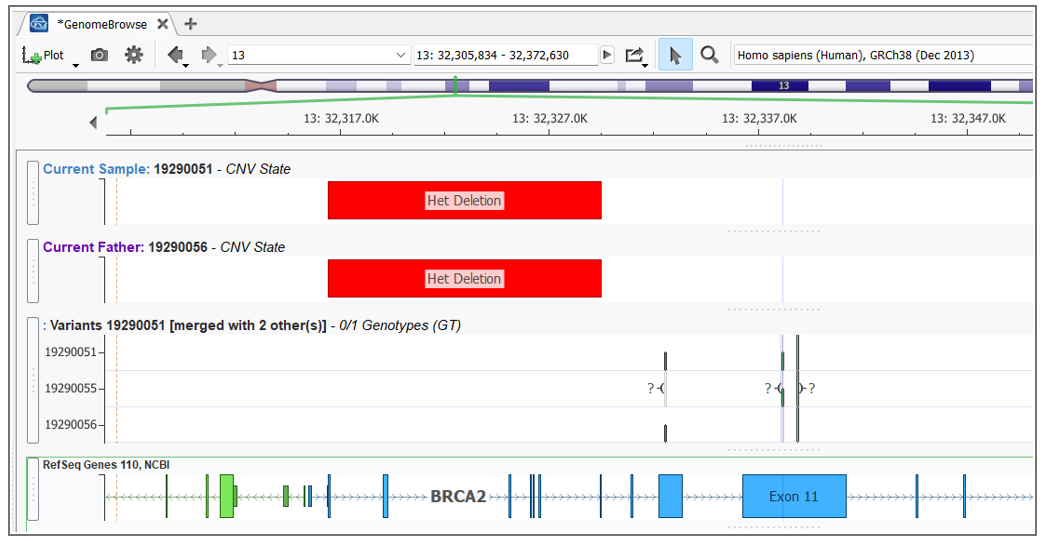
A real example: a proband inherits a BRCA2 SNP from their mother and a BRCA2 CNV (an exon-level deletion) from their father. Each parent is a carrier of a single variant and unaffected. The child, however, has no functional BRCA2 allele. In the GenomeBrowse window in VarSeq you can visualize both variant types in relation to each other — but without tooling that explicitly links them, this compound het pair would likely be reported as a single heterozygous finding, if it’s reported at all.
This is one of the most clinically significant sources of missed diagnoses in rare disease workups. A patient may receive a report noting a single heterozygous pathogenic variant in a recessive gene (technically accurate) while the CNV on the other allele goes undetected or unconnected.
CNV + CNV
Less common but entirely possible: both alleles carry structural deletions or duplications affecting the same gene. Detecting and interpreting two distinct CNVs in the same genomic region requires robust structural variant analysis and careful allele-level reasoning.
Why Cross-Class Compound Het Is Often Missed
The core problem is pipeline fragmentation. SNP/indel callers and CNV callers are separate tools, and their outputs are rarely analyzed together for compound heterozygous pairing. A lab may excel at finding SNPs and at finding CNVs, but if no step in the workflow asks “do these two different variant types affect the same gene on opposite alleles?”, the compound het will be invisible. According to research published in Genetics in Medicine, CNV-mediated compound heterozygosity accounts for a meaningful subset of undiagnosed recessive disease cases, particularly in genes where large deletions are a known mechanism.
Why Compound Heterozygosity Matters in Genetic Diagnostics
For any autosomal recessive condition, compound heterozygosity is just as disease-causing as homozygosity, but it arises through a different inheritance pattern and presents different detection challenges.
When two carrier parents have children, there is a 25% chance with each pregnancy that the child will inherit the pathogenic variant from each parent simultaneously. Carrier screening programs are built around this risk. But those programs are most effective when they account for the full spectrum of pathogenic variants in a gene, not just common SNPs.
Compound heterozygosity also explains phenotypes that simple heterozygosity cannot. If a patient presents with symptoms consistent with a recessive disorder, and sequencing finds only one heterozygous variant in the relevant gene, the clinical instinct may be to look elsewhere. But a second variant, particularly a CNV, on the opposite allele may be the missing piece. Incomplete variant detection has been documented as a major contributor to the diagnostic odyssey many rare disease patients experience.
Rare disease workups and reanalysis programs increasingly recognize this. Revisiting cases with expanded variant class detection — including CNVs, mobile element insertions, and mitochondrial variants — regularly yields diagnoses that were missed on initial analysis. Compound het across variant classes is frequently implicated.
For more on how variant interpretation fits into a comprehensive diagnostic workflow, see how VarSeq handles rare disease analysis and our overview of NGS variant filtering strategies.
How Compound Heterozygosity Is Detected in NGS Workflows
Detecting compound heterozygosity requires two things: finding both variants and correctly determining that they sit on opposite alleles (i.e., that they are in trans).
Phasing is the process of assigning variants to specific alleles. The gold standard is trio analysis — sequencing the proband alongside both parents. If a variant is found in the mother and a different variant in the father, and both are present in the proband, trans configuration can be inferred with high confidence. This approach works well for SNP + SNP pairs and, with the right tooling, for cross-class pairs as well.
Statistical phasing uses population haplotype data to infer phase without parental samples. It is less reliable for rare variants and cannot easily handle cross-class phasing between SNPs and CNVs.
Long Read Sequencing allow getting the phasing for an entire gene in a single NGS read, allowing a proband to have a compound heterozygous event detected even when parents are not available for a trio analysis. PacBio and ONT provide sequencing technologies that allow single reads to extended to 1,000 to 10,000 bases, providing clarity on whether two SNPs or a detected SNP and CNV are on the same allele.
The hardest cases are those involving variants from different detection pipelines. A SNP caller finds a heterozygous pathogenic missense variant. A CNV caller finds a heterozygous exon deletion in the same gene. Linking these two findings, confirming they affect different alleles, and surfacing them as a candidate compound het pair requires a workflow that integrates both variant classes into a unified analysis environment.
This is precisely where purpose-built tools make a difference. VarSeq is designed to detect compound heterozygous pairs across all variant classes, including SNP + CNV and CNV + CNV, within a single analytical environment, reducing the risk of cross-class compound hets being missed due to fragmented pipelines. For a detailed walkthrough of how this works in practice, Golden Helix has published a webcast — Combined Impact: New Tools to Assess Complex and Compound Heterozygous Variants with VarSeq — that covers the technical approach and shows the algorithm in action.
Putting It Together
Compound heterozygosity is not a rare edge case: it is a fundamental mechanism of recessive disease that every clinical genomics lab needs to account for. The SNP + SNP scenario is well understood, but the cross-class cases (SNP + CNV, CNV + CNV) remain a genuine blind spot in many workflows.
If your lab’s current pipeline detects variants in silos, some compound het diagnoses are being missed. Ensuring that your analysis environment can integrate variant classes and apply phasing logic across them is one of the highest-yield improvements you can make to diagnostic yield. Golden Helix’s VarSeq automates compound het detection across all variant classes, helping labs surface diagnoses that fragmented pipelines would leave behind.
Visit the VarSeq suite to see how it fits into your NGS workflow.
Frequently Asked Questions
What does compound heterozygous mean?
Compound heterozygous means a person carries two different pathogenic variants in the same gene, one on each copy of that gene (one from each parent). Both copies are disrupted, which can cause an autosomal recessive disorder even though neither variant is present on both alleles.
What is the difference between compound heterozygous and heterozygous?
A heterozygous individual has one normal allele and one altered allele, usually enough to prevent disease in recessive conditions. A compound heterozygous individual has two different altered alleles (one per copy of the gene), leaving no functional copy and typically resulting in disease.
Can compound heterozygosity be missed in genetic testing?
Yes, particularly when the two variants belong to different classes: for example, a point mutation on one allele and a copy number variant (CNV) on the other. If a lab’s pipeline analyzes SNPs and CNVs in separate workflows without cross-referencing them at the gene level, the compound het pair can go undetected.
Is compound heterozygosity the same as being a carrier?
No. A carrier is heterozygous for a single pathogenic variant and retains one functional allele. A compound heterozygous individual has no functional allele and is at risk for the full disease phenotype, not just carrier status.



