
Long-read sequencing technologies have opened new doors in genomics research, enabling more comprehensive analysis of structural variants, phasing, and complex genomic regions. To support these powerful applications, we’ve introduced several new features to GenomeBrowse that specifically enhance the visualization of long-read alignment files. These improvements are designed to help our users extract meaningful insights from their data while minimizing visual noise and maximizing clarity.
Split Alignments by Haplotype Phase
One of the most valuable aspects of long-read sequencing is its ability to phase variants across considerable genomic distances, providing crucial information about which variants occur together on the same chromosome. In alignment files, this phasing data is typically stored using the HP (Haplotype) tag which uses numeric identifiers (e.g. HP:i:1, HP:i:2) to specify the haplotype to which a read belongs.
GenomeBrowse’s new “Split by Phase” feature automatically groups reads based on their haplotype assignments. Instead of viewing a jumbled collection of overlapping reads, users can now see clear separation between haplotype groups, making it immediately apparent which variants occur together on the same chromosome copy. This visualization capability is particularly useful when validating phased genotypes from variant call files, enabling an quick assessment of the evidence supporting each haplotype assignment.

Stack by Base
While not all BAM files contain explicit phasing information, the long-read data itself often harbors implicit phasing relationships that can be identified by visualizing the reads supporting a specific mutation. GenomeBrowse’s new “Stack by Base” feature supports this use case by allowing users to reorder reads based on the nucleotide present at any specific genomic position.

This feature works by stacking reference-matching reads below the x-axis while positioning variant-supporting reads above it. The result is a powerful tool for discovering co-occurring variants even when formal phasing data is not available in the alignment file. Users can select any position of interest and immediately see which other variants tend to appear together on the same reads, effectively performing on-the-fly linkage analysis through visual inspection.
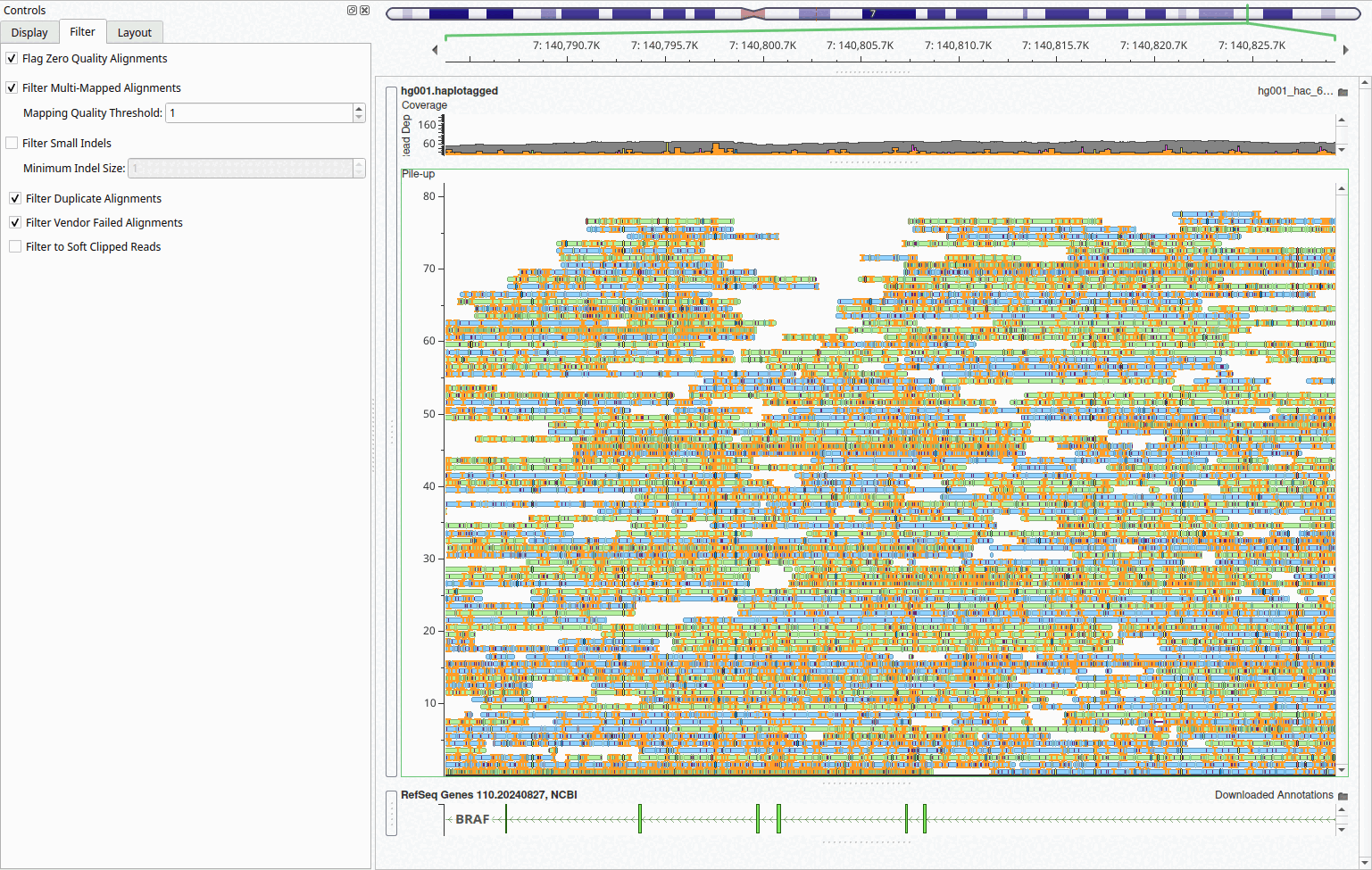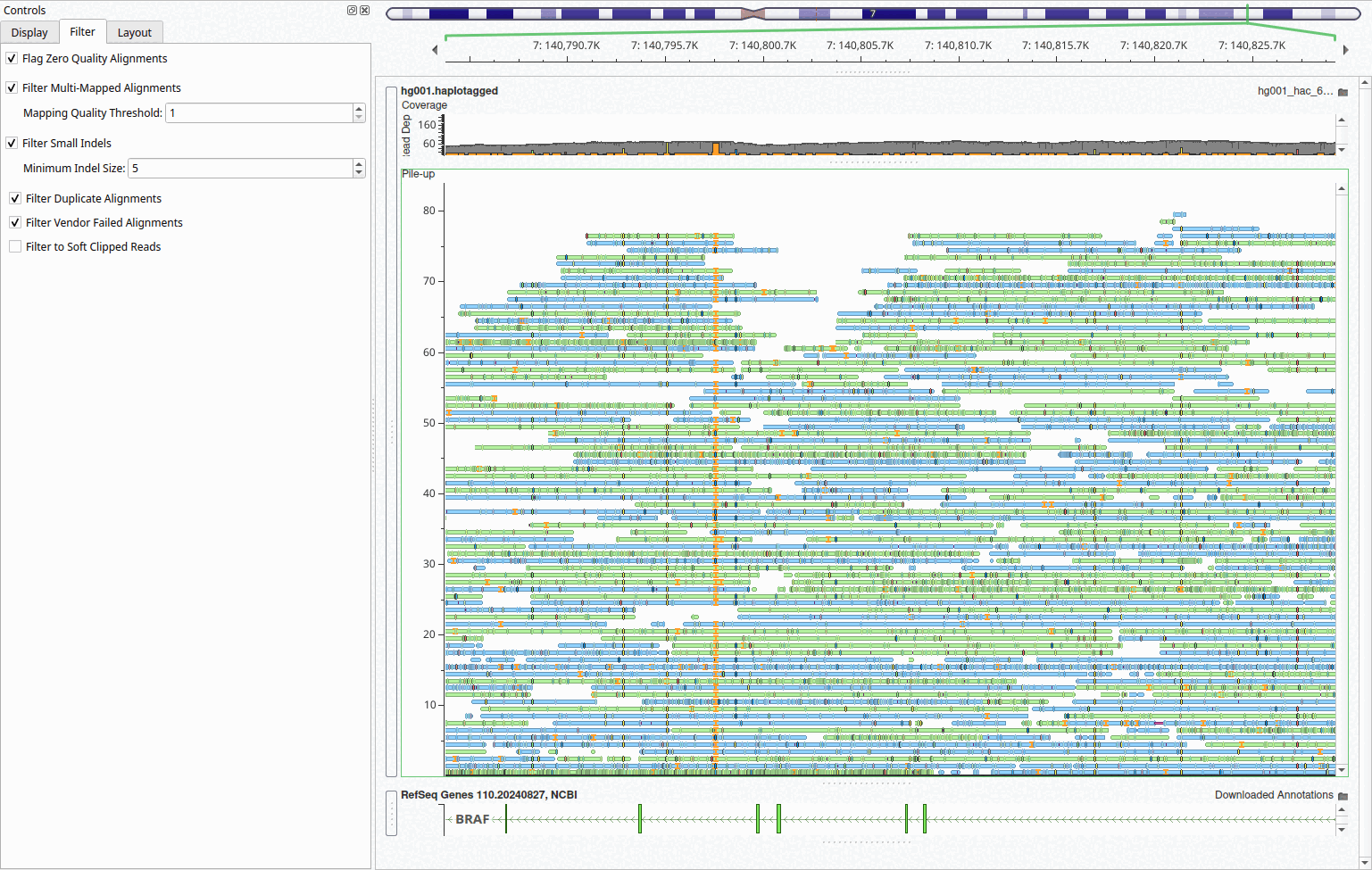
Filter Small Insertions and Deletions
When visualizing long-read sequencing data, the abundance of small insertions and deletions from sequencing artifacts can create visual clutter that obscures genuine variants. To address this issue, we have introduced a new feature to hide small insertions and deletions, allowing researchers to focus on larger, more confident variants while maintaining a cleaner view of read alignments and coverage patterns. This filtering capability is particularly useful when examining structural variants or assessing overall alignment quality, as the noise from technical artifacts can make it difficult to identify genuine mutations.
Conclusion
As long-read sequencing continues to become more widespread, tools that can effectively visualize and interpret this data become increasingly critical. These new capabilities enhance the utility of GenomeBrowse as a tool for long-read data analysis. The phasing capabilities help researchers leverage one of long-read sequencing’s greatest strengths: the ability to resolve complex genomic relationships over large genomic distances. Meanwhile, the ability to filter small insertions and deletions ensures that this rich information can be visualized despite the presence of artifacts that are commonly produced by long-read sequencing technologies.
Please contact our team if you would like to learn more about support for long read sequencing data in our software.




These updates seem like a huge improvement for long-read sequencing, particularly in terms of phasing. I can imagine this making it much easier to validate phased genotypes. Would be interesting to hear about its impact on the speed and accuracy of data analysis.