
If you spend enough time staring at VCF files, you eventually start to feel like you’re reading a secret language. Most of the time, things are straightforward with the Genotype (GT) representation: 0/0, 0/1, 1/1. Easy enough. But then one day, you import your VCFs for a trio analysis and see GT fields with ./., 0/., ./1, or even 1|0 staring back at you.
At first glance, it can feel like the VCF is trying to mess with you. In reality, these genotype representations are perfectly valid and they may show up when you’re working with family analysis such as a Trio or a large cohort that you often see with research projects. Fortunately, VarSeq is designed to handle these scenarios gracefully. Let’s decode a few of the usual GT suspects.
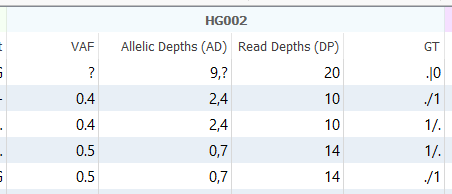
The Case of the Missing Genotype: ./.
The GT of ./. simply means that no genotype call was assigned for either allele in that sample. This representation often appears in cohort or trio analysis where a variant is present in one individual but not confidently detected in others. Rather than removing the variant entirely, the VCF keeps the record so the position can still be evaluated across all samples.
VarSeq preserves this structure during import so you can compare variants across the entire cohort, even when some samples don’t carry the variant. This is particularly helpful when searching for inheritance patterns in family analyses or exploring rare variants across large groups.
The Half-Missing Genotypes: 0/. and ./1
Sometimes you’ll see a GT where only one allele seems defined. For example:
- 0/. means the reference allele present and the second allele missing
- ./1 means that the alternate allele is present but the reference allele missing
These cases often appear when multi-allelic variants are split into multiple records during processing. The original site may have contained several alternate alleles, and when those are separated into individual rows, the GT representation can look a little unusual.
It may look strange at first, but it’s usually just a reflection of how the variant record was parsed, not an indication that something is wrong with the data. VarSeq has several ways of dealing with complex allele calls, which are covered here in this webinar.
Why Some Samples Have Depth and Others Don’t
Another common observation: one sample has depth metrics and allele counts, while another sample just shows missing values.
This happens because the variant may only have been called in one sample. When VarSeq imports a cohort VCF, it still lists the variant across all samples so you can analyze it collectively. The sample with the variant will have metrics like DP, GQ, or VAF, while the others will show missing values. In other words, the variant exists in the dataset, but not necessarily in every individual.
PASS… For Some Samples
You may also notice that not every sample meets the PASS filter for a given variant. This is often the result of single-sample variant calling pipelines where each sample is processed independently. When those results are combined into a cohort VCF, filtering outcomes can vary slightly from sample to sample.
If you prefer more consistent GT representation across a cohort or trio, joint calling can help. Joint calling analyzes all samples together, improving genotype consistency and often reducing missing calls.
Phasing: When the Slash Turns Into a Pipe
Occasionally the GT delimiter changes from a slash to a pipe: 1|0 instead of 0/1. That little vertical bar indicates phasing. In other words, the software knows which allele belongs to which chromosome copy. Phasing information is increasingly common in long-read sequencing datasets.
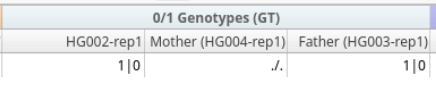
This is where VarSeq really shines. Using fields like Phased Set (PS), VarSeq can evaluate whether variants occur in cis or in trans, enabling powerful workflows such as compound heterozygous analysis in trios. Read more about Phasing in VarSeq here.
When in Doubt, Let VarSeq (and the FAS Team) Help
Large datasets, family analyses, and multi-allelic variants can produce genotype fields that look a little unconventional. But these representations are usually just the natural result of combining complex genomic data across multiple samples.
The good news is that VarSeq was built for exactly this kind of analysis, whether you’re working with trios, cohorts, or large sequencing studies.
And if a genotype still looks mysterious? The Golden Helix FAS team is always happy to help interpret what you’re seeing and make sure your analysis pipeline is working exactly the way it should. Because when the VCF gets weird, it’s nice to have experts, and great software, on your side. Contact [email protected] for more information today!



