
Short tandem repeats (STRs) are among the most challenging variants to characterize with traditional sequencing technologies. Many clinically significant STR expansions span hundreds or thousands of base pairs, far exceeding the read lengths of short-read platforms. As a result, countless disease-causing repeat expansions have historically gone undetected in genomic workflows, leaving patients without a molecular diagnosis.
Long-read sequencing technology offers a powerful solution to this problem. With reads long enough to span entire repeat regions, PacBio HiFi sequencing enables accurate genotyping, sequence composition profiling, and even methylation analysis of tandem repeats in a single assay. In this post, we will discuss how VarSeq and VSWarehouse bring these capabilities together into a seamless workflow, from STR genotyping to clinical reporting.
Genotyping Tandem Repeats with TRGT in VSWarehouse
VSWarehouse provides a PacBio Germline HiFi WGS Workflow, available for download from our Workflow Repository. Among its analysis stages is a dedicated step powered by TRGT, PacBio’s purpose-built tool for tandem repeat genotyping from HiFi data.
TRGT goes well beyond simple size-based genotyping. For each analyzed repeat, it profiles:
- Sequence composition: characterizing the exact repeat structure, including interruptions and motif variations.
- Mosaicism: detecting allele-level heterogeneity within the sample.
- CpG methylation: capturing epigenetic modifications directly from the long-read signal.
This depth of characterization is only possible because long reads span the full repeat region, providing context that short reads simply cannot offer.
Importing and Filtering STRs in VarSeq
The TRGT output can be imported directly into VarSeq alongside small variants, giving analysts a unified view of the sample’s genetic landscape. VarSeq supports annotation of STRs using STRchive, a curated resource of disease-associated tandem repeats. By leveraging STRchive, users can quickly identify clinically relevant repeat expansions and incorporate them into clinical reports.
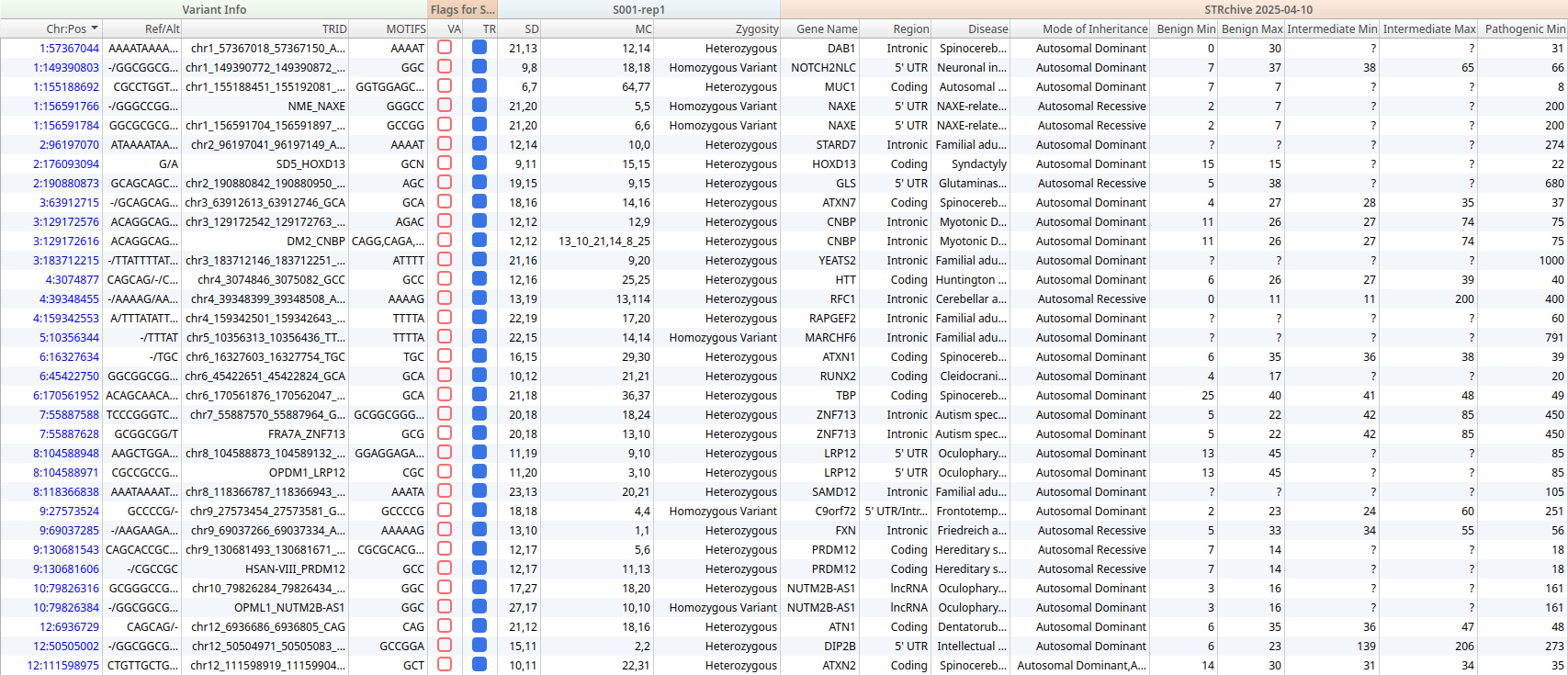
Using a dedicated STR filter chain we can identify variants with a valid repeat information that overlap repeat regions in the STRchive annotation source. In the example dataset shown here, filtering revealed 56 STRs with disease-association information in STRchive. These filtered variants were added to an STR record set, flagging them for inclusion in the clinical report alongside any small variant findings.
Clinical Reporting of STR Findings
Once filtering and annotation are complete, VarSeq makes it straightforward to produce a clinical report using the STR Report Template shipped with the software.
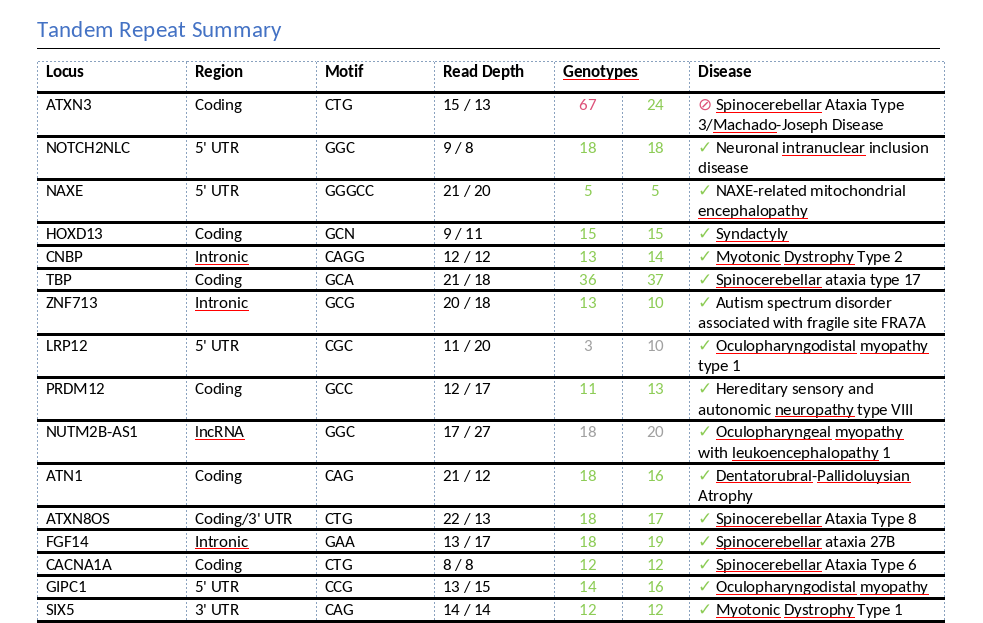
Within the generated report, the Tandem Repeat Summary section provides a clear overview of clinically relevant findings. In this example, the sample contains a heterozygous pathogenic repeat expansion in the ATXN3 gene, which is associated with Machado-Joseph Disease, an autosomal dominant neurodegenerative disorder.
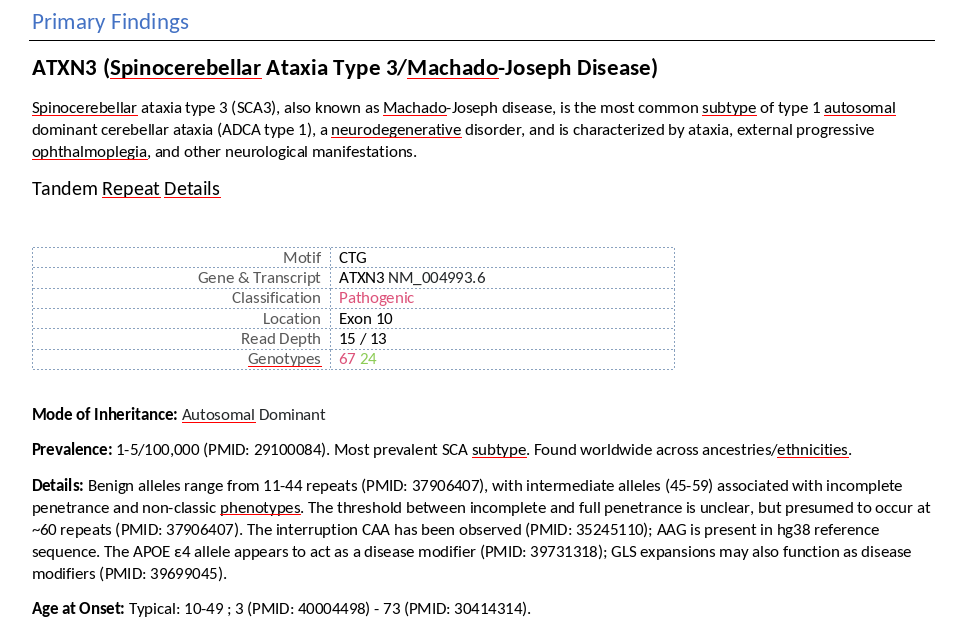
Scrolling further into the report, the Primary Findings section provides detailed clinical context for each pathogenic STR, including Mode of Inheritance, Prevalence, and Age at Onset for the associated disease. These details are pulled directly from STRchive and include inline citations linking to relevant literature, supporting transparent and evidence-based reporting.
While the shipped STR report template provides an excellent starting point, both the underlying report-generation code and the Microsoft Word template used to format the output are fully customizable. This flexibility allows laboratories to tailor the content, layout, and styling of their reports to meet specific institutional requirements and branding preferences.
Conclusion
Long-read sequencing enables a level of resolution for tandem repeat analysis that is difficult to achieve with short-read technologies. By capturing full repeat expansions within single reads and preserving sequence context, long-read data allows for more accurate genotyping and more-comprehensive biological insights.
For patients with neurological, neuromuscular, or other conditions potentially driven by repeat expansions, this translates directly into improved diagnostic yield. By combining TRGT genotyping in VSWarehouse with the annotation, filtering, and reporting capabilities of VarSeq, we’ve built a workflow designed to make STRs clinically actionable, from raw reads to a finalized clinical report. To learn more about support for STR analysis in VarSeq and VSWarehouse, please reach out to our team.



