Getting the Most from Assessment Catalogs in VSWarehouse

One of the most frequent questions from our customers is some version of: “How do we keep our team from re-evaluating the same variant every time it shows up in a new sample?” The answer, in nearly every case, is assessment catalogs and more specifically, learning to use them strategically.
This post walks through what assessment catalogs are, the different ways labs are putting them to work, and how VSWarehouse makes them queryable across your entire sample history.
What is an assessment catalog?
At its core, an assessment catalog is a curated collection of records tied to a specific data type: variants, CNVs, SVs, regions, or samples that your team builds and maintains over time. Unlike static reference databases, these catalogs live inside your VSWarehouse workspace and reflect your lab’s own clinical experience and interpretation decisions.
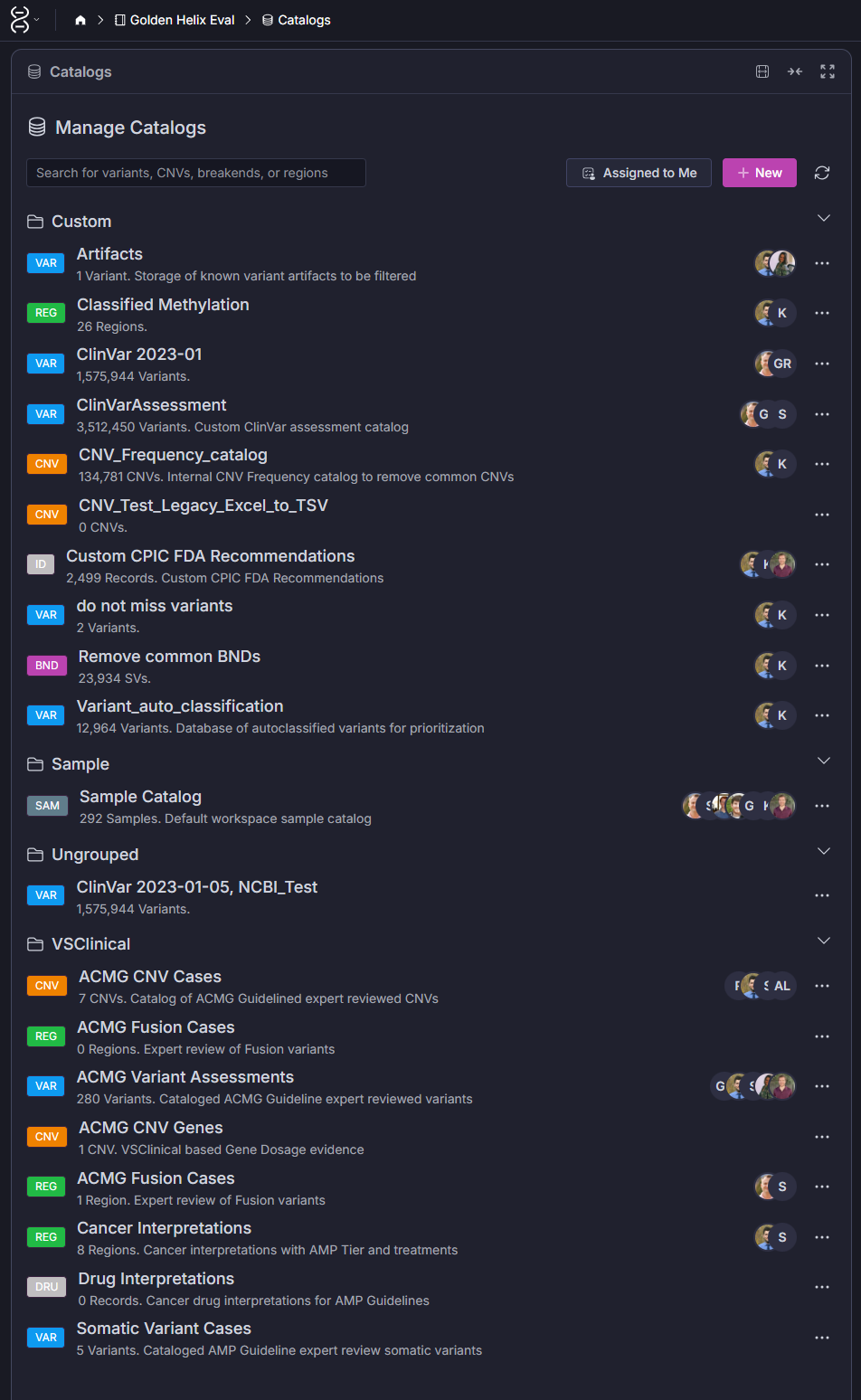
VSWarehouse supports several catalog types including ACMG Variant Assessments, Somatic Variant Cases, and Cancer Interpretations. But the real power comes from the custom catalogs your lab builds on top of those. (Screenshot 1)
Four ways labs use assessment catalogs
The most productive VSWarehouse users typically have catalogs serving at least three or four distinct functions. Here’s how to think about each one.
1. Storing classifications and interpretations
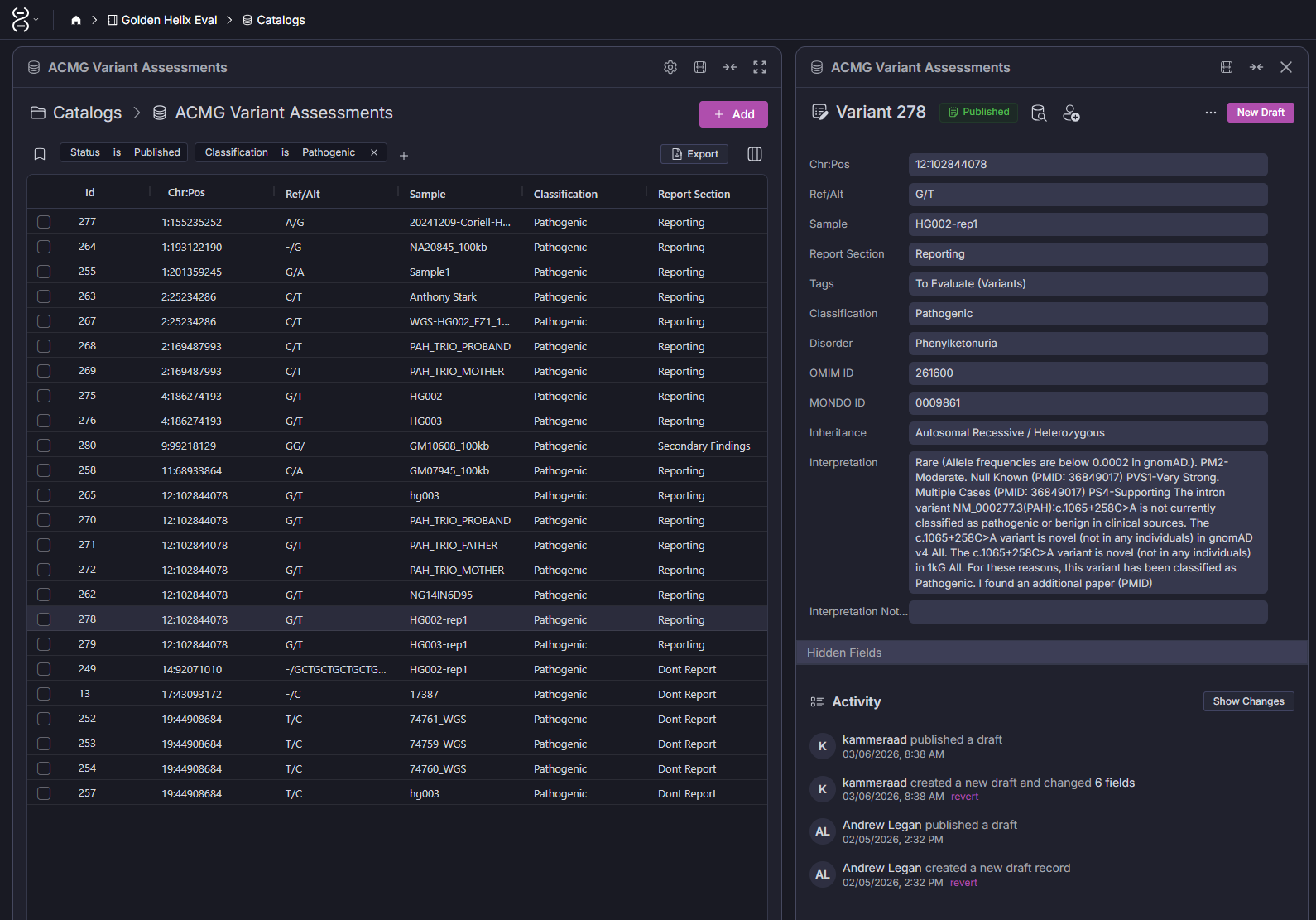
This is the most foundational use case. When a variant is classified in VarSeq, let’s say, a pathogenic PAH variant identified in a phenylketonuria case, that classification and its associated interpretation can be written back to the ACMG Variant Assessments catalog. The next time that same variant appears, VarSeq pulls the existing classification automatically, including the supporting evidence summary your scientist wrote the first time around.
The result is a compounding return on the time your team spends on variant review. The catalog entry is versioned (draft → published), tied to the sample and disorder it was first observed in, and linked to the specific OMIM/MONDO identifiers your lab uses. As shown in the Screenshot 2, you can see the full interpretation thread including activity history, who published the draft, and when.
2. Querying across samples in VSWarehouse
This is where assessment catalogs shift from being a storage system to becoming a knowledge base you can interrogate. From the Catalogs interface in VSWarehouse, any classified variant can be searched across the full sample history — not just to review its classification, but to instantly answer questions like: Which other samples carry this variant? What were their disorders and inheritance patterns? What did the sample catalog record for their clinical context?
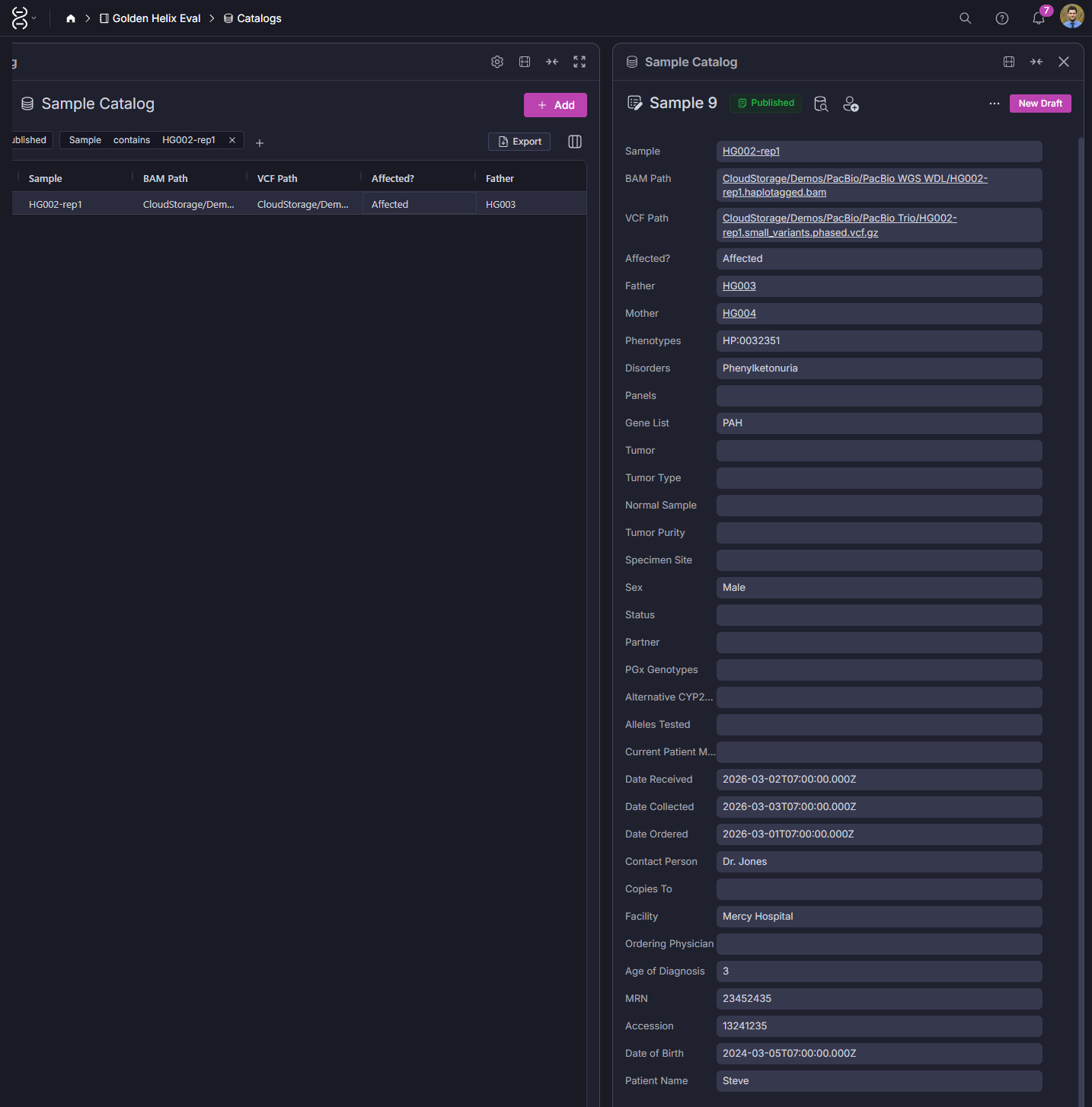
The sample catalog record seen in screenshot 3 captures the full clinical context for each sample: BAM and VCF paths, pedigree relationships, phenotypes, disorder, ordering physician, facility, MRN, and accession number. When you navigate to a variant in the assessment catalog, you can trace directly back to the sample records of everyone who carries it. That connection is what turns a variant database into a clinical knowledge system.
3. Filtering known artifacts
Low-quality variants that appear consistently across samples as sequencing artifacts tied to your library prep chemistry, platform-specific noise, or false positive indels for example and can be cataloged in a dedicated Artifacts catalog. Once a variant is added, VarSeq filters can automatically exclude it from all future projects without requiring a scientist to touch it. One entry, permanent benefit.
This becomes especially critical with Structural Variants that are a substantial fraction of platform-specific noise that simply doesn’t exist in public databases like gnomAD.
4. Removing common CNVs and SVs
Genome-wide CNV and SV analysis generates an enormous noise problem. Common copy number variants, particularly in low-complexity regions, appear in the general population at high frequency and are clinically irrelevant in most contexts, but they flood variant lists and slow review. The CNV_Frequency_catalog visible in the Catalogs view (Screenshot 1) addresses exactly this: a curated internal frequency reference that reflects what’s common in your specific patient population and sequencing workflow, not just what’s cataloged in public cohorts.
What this looks like in practice
The capabilities above are demonstrated in detail in the Golden Helix webcast Next-Gen Sequencing at Scale: VSWarehouse, which walks through multi-sample workflows, catalog-based queries, and the VarSeq integration pipeline that writes classifications back to the warehouse automatically. If your team hasn’t watched it, it’s worth an hour, the live demo of navigating from a classified variant back through sample history is particularly useful context for new users.
Getting started
If you’re setting up or evaluating VSWarehouse and want to see this in action with your own data type, whether that’s a gene panel, whole exome, whole genome, or long-read workflow, please reach out to our FAS team. We can walk through catalog setup, query workflows, and how the sample catalog amplify your workflow efficiency and structure maps to your existing LIMS or EHR.