In the September 2021 monthly update to our curated ClinVar track, we made some changes that will result in roughly another 7,000 Likely Pathogenic and Pathogenic variants being available for variant annotation and use in the ACMG auto-classification system.
Consensus Between Labs
ClinVar has nearly one million unique variant classification records that are curated into multiple annotation tracks used in VarSeq and VSClinical on a monthly basis. Clinical labs in the US and across the globe contribute their own variant classifications through a submission process that includes the following key properties:
- Interpretation: The ACMG classification determined by the labs, usually following the 5-tier ACMG system
- Condition: The disorder evaluated during the interpretation process
- Supporting Information: Optional additional information, including full interpretation text, the HGVS of the variant as the lab described it etc.
When multiple labs have submissions for a single variant, ClinVar summarizes the submissions and if they agree provides a clear interpretation such as “Pathogenic” or “Benign” is provided for the variant.
But when individual lab submissions disagree, ClinVar does not try to perform a “majority vote” or run a consensus algorithm. It simply sets the Interpretation field to something like “Conflicting interpretations of pathogenicity”. A good example of this is NM_032588.3(TRIM63):c.739C>T (p.Gln247Ter), which Invitae has submitted with an Interpretation of Likely Benign, while Ambry submitted as Pathogenic. Certainly, this variant represents a rare case in the ClinVar database but does exemplify the possibility of disagreement at the variant level. ClinVar is simply an aggregator of individual lab submissions.
Conflicting versus Imperfect Consensus
The VarSeq curation of ClinVar improves upon this wide range of values in the “Interpretation” field by creating a “Classification” field that consists of the 5 ACMG classification states of Pathogenic, Likely Pathogenic, Uncertain Significance, Likely Benign and Benign with the additional categories of Conflicting, Association Not Found and Other. The Classification field “cleans up” the wide range of variants cataloged in ClinVar. In VarSeq this field, with its fixed set of values, is often used for filtering. In VSClinical it informs the ACMG Auto-Classifier of previous clinical assessments and is used in the recommendation engine for various benign and pathogenic criteria.
We have recently had several customers note that there are several variants that are currently listed as “Conflicting” that are, in fact, well-established Pathogenic variants for specific diseases.
An example of this is NM_000410.3(HFE):c.845G>A (p.Cys282Tyr), a variant well-established in hereditary hemochromatosis and explicitly listed as a known pathogenic variant in disease-specific guidelines. Yet, the ClinVar page for this variant lists the interpretation as Conflicting interpretations of pathogenicity. Clicking on this variant in GenomeBrowse pulls up the variant track details:
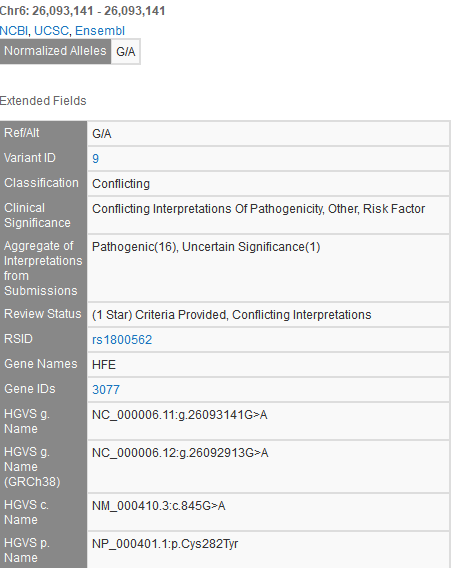
The VarSeq annotation “Aggregate of Interpretations from Submissions” field provides a convenient roll-up of the individual lab submission “Interpretation” values. As you can see, there are 16 individual lab submissions of this variant being Pathogenic, and a single lab submission of it being Uncertain Significance. The following is a representative excerpt from Illumina’s lab submissions supporting information:
The HFE c.845G>A (p.Cys282Tyr) missense variant is one of the two most common and well-studied pathogenic variants associated with hereditary hemochromatosis (HH), with approximately 80-87% of HH type 1 patients of European origin being homozygous or compound heterozygous for this variant (Feder et al. 1996; Gallego et al. 2015; Press et al. 2016).
But because of the single “Uncertain” submission for this variant, it is unfortunately marked as “Conflicting” by ClinVar and thus in our own ClinVar tracks. That is until this month.
Changing Conflicting to Pathogenic or Likely Pathogenic
After doing some analysis and discussing with our users, we made the decision to update our curation of ClinVar to change the creating of the Classification field to have specific logic to handle these variants. As the two examples above demonstrate, simply looking at ClinVar’s own Interpretation summary field is insufficient to differentiate the case of true conflicting submissions and a disagreement along with the severity of pathogenicity.
To this end, we have established a heuristic that as long as there are no submissions of Benign or Likely Benign evidence, the Classification field will take the highest submitted classification of Pathogenic or Likely Pathogenic.
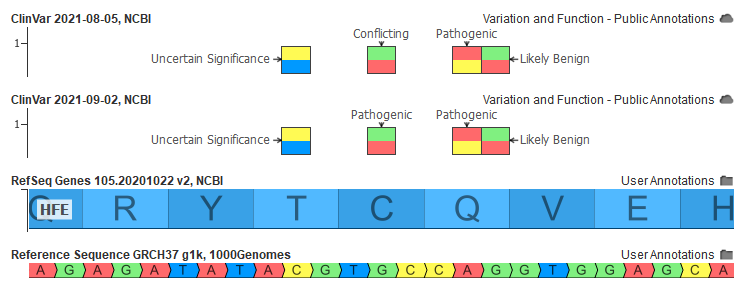
With this change in place, the September ClinVar now has about 7,000 more Pathogenic and Likely Pathogenic variants that were previously set as Conflicting. Transforming and optimizing the raw ClinVar data to prepare it for supporting the clinical genomics interpretation work of VarSeq and VSClinical requires constant vigilance and ongoing investment. This is one of the many reasons we see labs adopting VarSeq and VSClinical. If you have questions about how a variant is curated or have further suggestions on other variant edge-cases to investigate, please contact us!



