
One-Click Secondary Analysis in VSWarehouse
A child with an undiagnosed condition has, on average, seen seven specialists and waited more than four years before receiving a genetic diagnosis. A cancer patient waiting on somatic variant results to determine whether an EGFR-targeted therapy is appropriate sits in treatment limbo measured in weeks. A pregnant couple asking about their carrier status for a rare autosomal recessive condition needs an answer before a major reproductive decision. Despite the increasing use of clinical genetics in routine practice, many institutions lack the computational power and integration necessary to handle many samples reliably, rapidly, and at scale.
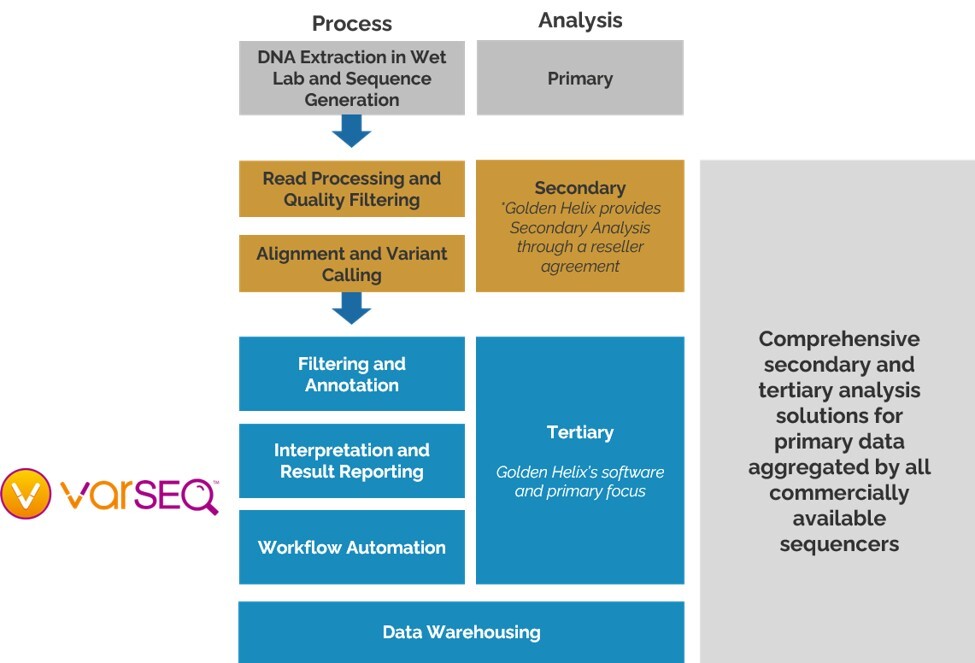
Clinical genetics workflows are broken into three stages: 1) Primary analysis where sequences are generated, 2) Secondary analysis where reads are processed and variants are called, and 3) Tertiary analysis where clinically important variants are separated from mundane variants and clinical reports are generated (Figure 1). While long-read sequencing technologies are gaining widespread adoption, short-read sequencing remains the dominant platform in clinical workflows.
What is Short-Read Sequencing?
Short-read sequencing – the class of technology that includes Illumina platforms – works by fragmenting a DNA sample into millions of small pieces, typically paired 75-150 bases in length, then reading each fragment from both ends simultaneously. The result is a FASTQ file: A list of millions of short reads, each paired with a quality score estimating the confidence of each individual base call. Short-read platforms have become the workhorse of clinical genomics because they are fast, highly accurate at the single-base level, and cost-effective at scale – a whole exome can be sequenced to clinical depth in a matter of hours. The limitation is inherent to the approach: each read captures only a small window of the genome. The reads do not arrive in genomic order, and low complexity and repetitive regions produce reads that look nearly identical to reads from elsewhere. Reassembling this data in a coherent, clinically interpretable picture of a patient’s genome is the job of secondary analysis. At clinical scale, secondary analysis is computationally expensive and difficult to streamline/organize into subsequent tertiary analyses. This is where most labs hit their operational ceiling.
Why is secondary analysis difficult to scale and automate?
Secondary analysis is the computational bridge between raw instrument data and clinical variant calls. It does not interpret variants – that is tertiary analysis which tools like VarSeq perform. What it does is transform millions of disordered short reads into a clean, well-characterized map of a patient’s genome relative to a reference, with every difference flagged, quantified, and quality-scored. Golden Helix tertiary analysis is agnostic of secondary analysis inputs, allowing users full customization of their workflows.
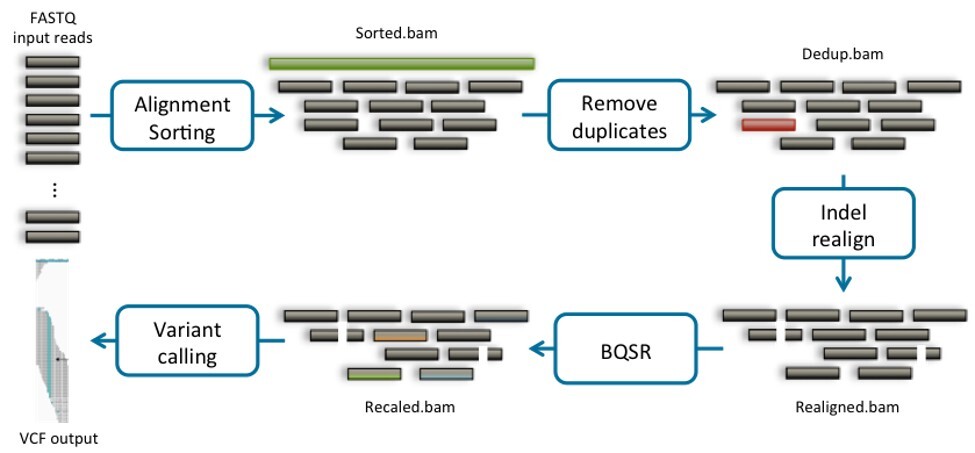
The process runs in four stages (Figure 2 which details secondary analysis with Sentieon). First, alignment: each short read is mapped back to the reference genome using a tool like BWA-MEM. The aligner finds the position in the three-billion-base reference where each 150-base read most likely originated, assigns a mapping quality score, and writes the result to a BAM file. Reads from highly repetitive genomic regions may map equally well to multiple locations; these mulimappers are flagged rather than placed, because placing them incorrectly would introduce false variant calls. Second, the alignment is sorted a deduplicated. PCR amplification during library preparation creates multiple identical copies of the same original DNA fragment. If those duplicates enter the variant calling step, they inappropriately inflate evidence for a variant when they are in fact artifacts of a single sequencing event. Deduplication removes this inflation and produces a cleaner estimate of true allele frequency.
Third, base quality score recalibration (BQSR) corrects systematic biases in the sequencer’s own confidence estimates. Sequencers report a quality score for every base they call, but those scores carry platform-specific biases – overconfident in some sequence contexts, underconfident in others. BQSR models those biases using a set of known variant sites and adjusts the scores accordingly, which directly reduces false positive variant calls downstream. Sentieon’s QualCal implements this step, and their DNAscope called extends the principle further by applying a deep learning model trained on high-confidence genomic benchmarks to improve variant call accuracy beyond what traditional statistical approaches achieve – particularly in challenging sequence contexts. Most modern secondary analysis pipelines offer equivalent recalibration steps, all of which VSWarehouse supports as first-class pipeline components.
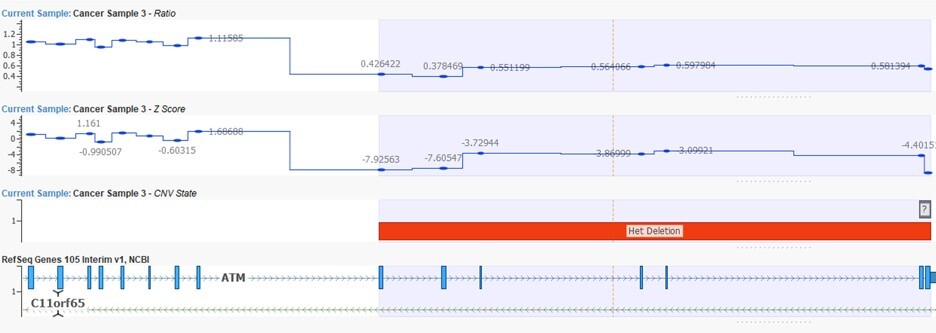
Fourth, variant calling. SNVs and small insertions or deletions are called from recalibrated BAM using a haplotype-aware caller that considers the local assembly of reads around each candidate site. The output is typically a VCF: a structured list of every position that differs from the reference, annotated with allele frequency, genotype, and per-call quality metrics. Copy number variants are called separately, from depth-of-coverage data in the BAM rather than from individual base calls. Coverage across each target region is compared against a reference panel of normal samples; regions with statistically significant gain or loss are flagged as CNVs, with configurable thresholds for depth and coverage variability. While VarSeq is primarily a tertiary analysis tool, it has its own integrated CNV caller (Figure 3).
At the end of secondary analysis, the lab has two core output types: the BAM file (reads aligned to the reference genome) and the VCF, the called variants ready for tertiary interpretation. Secondary analysis cannot tell you which of those variants matters clinically. That requires annotation, filtering, and clinical curation. But it cannot happen without accurate, reproducible secondary output. Everything downstream depends on the quality of the alignment and variant calls. When processing hundreds or thousands of samples, all the secondary analysis steps need to be reproducible, highly documented, properly organized/formatted, and configurable to integrate into tertiary analysis. Running large numbers of samples individually is virtually impossible without additional automation and organization software.
Containerization, Automation, and VSWarehouse
The difficulty in integration and scaling isn’t a shortcoming of the tools, but an operational obstacle. The tools for short-read secondary analysis are well-established and highly capable. What breaks down at scale is everything around them: managing which version of each tool was run on which sample, ensuring that a pipeline executed today produces the same result if re-run six months from now, making that pipeline accessible to analysts who are not comfortable on the command line, and producing output that flows cleanly into downstream interpretation without manual reformatting or intervention. These are infrastructure problems.
VSWarehouse ships with pre-bundled, validated secondary analysis pipelines ready to run on day one, no configuration or IT overhead required. Every pipeline runs in an isolated, version-locked environment that behaves identically across deployments, whether cloud, on-prem, or air-gapped. For regulated laboratories, this means a validated, auditable process out of the box. For research programs spanning months or years, it means results that stay comparable over time and protecting the integrity of your data as tools and protocols evolve.
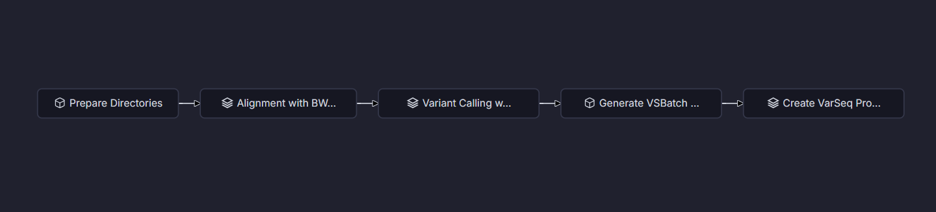
VSWarehouse transforms secondary analysis from a manual bioinformatics task into a turnkey, automated workflow. Labs configure a pipeline once: VSWarehouse handles execution, monitoring, provenance capture, and structured output delivery. Analysts focus on results, not infrastructure. VSWarehouse is pipeline-agnostic. Golden Helix partners with Sentieon as our recommended high-performance engine, but labs running GATK, DRAGEN, or in-house workflows plug in seamlessly and gain the same automation and compliance benefits out of the box. Figure 4 shows an example workflow that takes FASTQ files through alignment, secondary analysis, and generates a VarSeq project, a multitude of actions all done with one click.
The operational improvements this delivers address each of the core scaling challenges directly:
- Compute at scale. VSWarehouse orchestrates job scheduling across cloud, on-prem, and air-gapped deployments.
- Reproducibility. Container-locked pipeline definitions guarantee the same tool versions run on every sample, every time.
- Compliance documentation. Every run automatically logs tool versions, parameters, input checksums, and outputs. No manual documentation required.
- Cross-site standardization. A single shared pipeline definition enforces identical workflows across all sites, making multi-site QC and cohort analysis tractable.
Limitations of Short-Read Secondary Analysis
While short-read variant calling forms the bedrock of modern clinical diagnostics, it has significant shortcomings. At 150 base pairs per read, short-read platforms struggle to characterize structural variants, cannot reliably sequence through repetitive regions and homologous gene families like SMN1/SMN2, and cannot determine whether two variants on the same gene sit on the same chromosome or opposite ones. This is a critical distinction that is often the difference between a carrier and an affected patient (compound heterozygosity).
These limitations are inherent. to short-read technology and cannot be overcome by improvements in alignments or variant calls. That is where long-read sequencing comes in. In Part 2 of this series, we look at how reads measured in kilobases rather than base pairs change what secondary analysis can see, how VSWarehouse standardizes and streamlines long-read sequencing and its associated information (e.g., phase genotypes), and what that means for the clinical cases where short-read sequencing currently leaves questions unanswered.
Short-read secondary analysis is a mature, well-validated process that underpins the majority of clinical genomic testing today. The bottleneck in clinical genomics is the operational infrastructure required to run those tools reproducibly, at scale, and in compliance with regulatory standards. VSWarehouse provides the infrastructure to overcome these problems. VSWarehouse transforms secondary analysis from a manual bioinformatics task into an automated, documented, and reproducible point-and-click workflow. VSWarehouse is pipeline-agnostic: whether a lab runs Sentieon, GATK, DRAGEN, or an in-house workflow, the same containerization, automation, and compliance infrastructure applies.



