
Family-based analysis is one of the most powerful tools in rare disease genomics. When multiple relatives are affected, investigators naturally look for a shared genetic explanation, often with good reason. Shared inheritance patterns frequently provide the critical clue needed to identify a causal variant.
The case described here is a hypothetical scenario constructed with fictional patient details to protect privacy, but it is inspired by a real multi-affected family encountered during rare disease analysis. The diagnostic pattern itself is genuine: two affected siblings, each carrying a distinct de novo pathogenic variant in an unrelated gene. While uncommon, such cases challenge one of the most deeply ingrained assumptions in clinical genetics: that affected family members necessarily share the same genetic etiology.
In this post, we demonstrate how VarSeq’s variant prioritization and ACMG classification independently identified the molecular diagnosis for each sibling. More importantly, we show why evaluating each patient on the basis of their own phenotypic profile, rather than forcing a shared explanation, was essential to reaching the correct diagnosis.
The Case: Two Siblings, Two Independent Diagnoses
The proband is a young child presenting with infantile spasms, hypotonia, global developmental delay, hypsarrhythmia, and intellectual disability. This is a severe combination of early-onset neurodevelopmental phenotypes with a presentation strongly suggestive of epileptic encephalopathy. The affected sibling, by contrast, presents with macrocephaly, coarse facial features, mild intellectual disability, hearing impairment, and abnormal heart morphology, a dysmorphic syndromic profile with no meaningful clinical overlap with the proband.
The natural clinical instinct when two siblings are affected is to search for a shared genetic explanation: a recessive variant inherited by both siblings from carrier parents, or a dominant variant with variable expressivity. That instinct is well-founded in most cases, and it is built into the structure of most family-based variant analysis workflows. Here, however, it would have led the analysis in entirely the wrong direction. The two children are affected for completely independent reasons, and identifying that required the analytical framework to evaluate each patient on their own terms.
Variant Prioritization: How VarSeq Navigated the Complexity
Proband Analysis
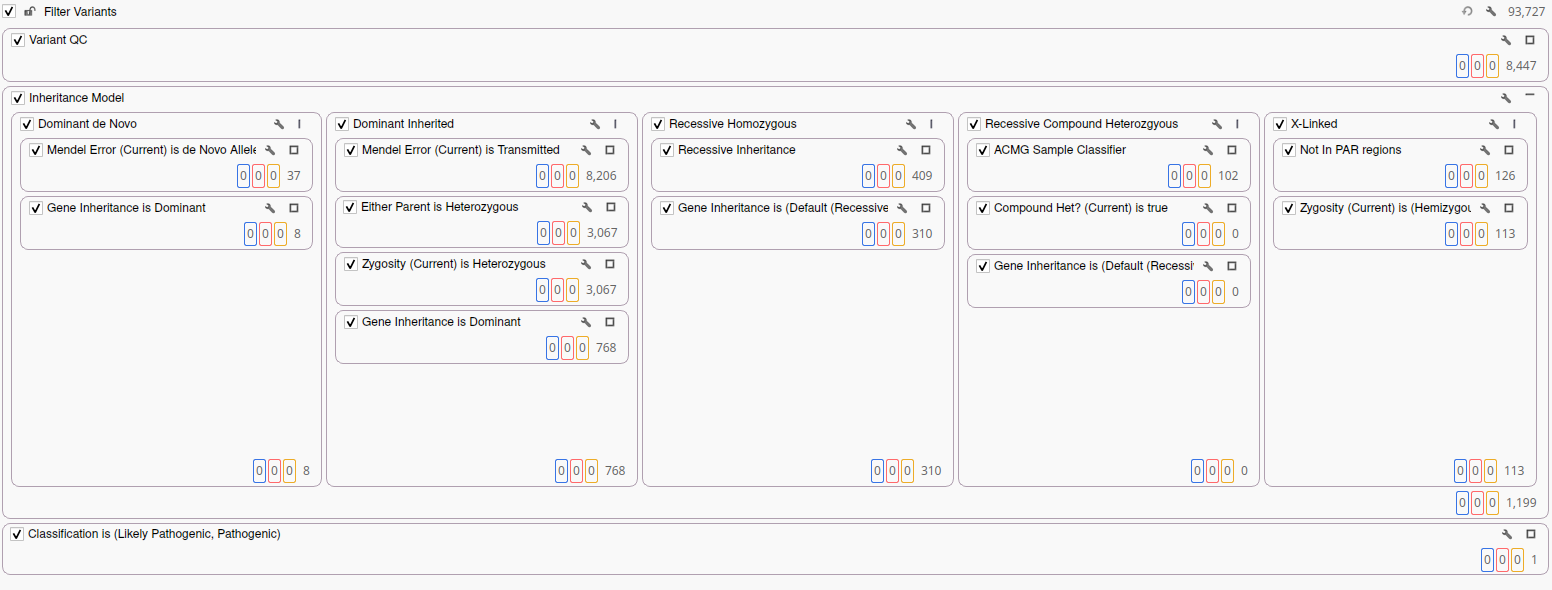
The first step in our analysis is the construction of an inheritance-aware filter chain that partitions variants by the mode of inheritance they are compatible with. Parallel filters evaluate de novo variants, dominant inherited variants, recessive homozygous variants, recessive compound heterozygous pairs, and X-linked variants simultaneously. This ensures that no compatible mode of inheritance is overlooked while keeping the analysis structured. After applying these filters, we are left with over 1,000 candidate variants across all inheritance categories, a number far too large for efficient manual review.
To reduce this candidate set to a clinically actionable list, we apply VarSeq’s automated ACMG Sample Classifier. This algorithm evaluates each variant based on the ACMG guidelines applying criteria systematically based on variant type, population frequency, computational predictions, and available clinical evidence. The classifier identifies variants meeting the threshold for Pathogenic or Likely Pathogenic classification and flags them for review. Applying the ACMG Classification filter to the proband’s candidate set yields a single Pathogenic variant: SCN2A p.Arg102Ter.
In the VarSeq variant table, the picture comes together quickly. The variant is a heterozygous de novo stop-gained mutation. This is a high-confidence loss-of-function call in a well-established dominant disease gene. Alongside the variant, VarSeq displays the PhoRank gene ranking score, which reflects the degree of phenotypic match between the patient’s HPO terms and the gene’s known phenotype associations. For SCN2A in this proband, PhoRank returns a gene rank above 98%, reflecting the near-perfect alignment between the patient’s phenotypes (infantile spasms, hypotonia, hypsarrhythmia, global developmental delay, and intellectual disability) and the established clinical features of SCN2A-related Early Infantile Epileptic Encephalopathy. Importantly, the variant is absent from the sibling entirely, which begins to undermine the shared-etiology hypothesis before the sibling’s analysis has even begun.

Analyzing the variant in VSClinical, the ACMG criteria supporting the Pathogenic classification are displayed alongside the reasoning behind their application.

A review of ClinVar assessments confirms that this exact variant (SCN2A c.304C>T) has been previously classified as Pathogenic and has been observed in multiple individuals with clinical features of Early Infantile Epileptic Encephalopathy 11.

This prior clinical observation provides strong independent support for pathogenicity and, combined with the excellent phenotypic match in PhoRank along with the de novo inheritance, makes a compelling case for reporting this as the causal variant. The absence of this variant in the sibling is not a weakness of the interpretation, since it is consistent with the proband’s phenotypes being entirely absent in the sibling, and it is concrete evidence that the two conditions may have independent genetic causes.
Sibling Analysis
The sibling’s analysis follows the same workflow. The inheritance-aware filter chain generates a candidate variant set, and the ACMG auto-classifier reduces it to a single Pathogenic variant: CHD4 p.Arg1068His. Like the proband’s variant, this is a heterozygous de novo missense mutation in an autosomal dominant disease gene. CHD4 encodes the catalytic subunit of the nucleosome remodeling and deacetylase (NuRD) complex, and heterozygous de novo variants cause Sifrim-Hitz-Weiss syndrome, which is characterized by macrocephaly, coarse facial features, intellectual disability, hearing impairment, and structural cardiac anomalies. PhoRank ranks CHD4 above 99% for the sibling’s phenotype set, reflecting the same near-perfect alignment seen in the proband’s analysis.

Conclusion
In a multi-affected family, the gravitational pull toward a shared genetic explanation is usually justified. Most workflows are structured, implicitly or explicitly, to prioritize variants that segregate across affected family members. In this case, that approach would have deprioritized both causal variants in favor of any variant the two siblings happened to share, regardless of how well it matched either patient’s phenotype.
What made the difference here was that VarSeq’s phenotype-driven ranking evaluated each sibling independently against their own HPO term set. The proband’s phenotype profile (dominated by epileptic encephalopathy features) and the sibling’s profile (dominated by dysmorphic syndromic features) are so clinically distinct that ranking on broad terms like “neurodevelopmental disorder” would have treated them as similar patients and potentially converged on a shared variant as the preferred explanation, despite the phenotypic differences between the siblings.
The result is a case that illustrates something important about the limits of family-based assumptions in variant prioritization: shared family membership is evidence of shared genetic etiology only when the phenotypes are also shared. When they are not, the analysis has to be willing to let each patient tell their own story and the software infrastructure has to be built to support that. To learn more about family-based variant analysis in VarSeq, including inheritance-aware filtering, phenotype-driven prioritization, and automated ACMG classification, please reach out to our team.


