
Somewhere in your last sequencing run is a variant that nobody has ever seen before. It might be the answer to a patient’s decade-long diagnostic odyssey, or it might be background noise. Variant classification is how you tell the difference. It is the step where a row in a VCF becomes a clinical answer.
For ten years, the rulebook for making that call has been the 2015 ACMG/AMP guidelines. What fewer people have tracked is how thoroughly that rulebook has been rewritten since. The headline framework is the same one labs adopted a decade ago, but underneath it, ClinGen has replaced, recalibrated, or retired most of the original criteria. It happened quietly enough that a pipeline built in 2016 and never touched since is now scoring variants by rules the field has moved on from. Meanwhile, cancer interpretation grew up alongside it, with a parallel framework for oncogenicity that borrows ACMG’s structure but answers a completely different question.
I covered all of this in our recent webinar, Modernized ACMG and Cancer Variant Classification: Implementing the Latest ClinGen Guidance. This post is the written version: what changed, why it changed, and how a single platform can run both the germline and the cancer frameworks off the same modernized evidence. The example cases are the ones I walked through live in the demo.
What Is ACMG Variant Classification?
Every clinical genome turns up variants that have never been reported in a patient. Classification is the discipline of deciding, on the basis of evidence, what each one means for that person and their disease. The goal isn’t just an answer. It is a reproducible answer. Hand the same variant to two analysts working from the same framework, and they should land in the same place. When they don’t, the framework is not providing enough guidence.
The 2015 ACMG/AMP guidelines (Richards et al., Genetics in Medicine) provided the field with a framework, built on a two-step process: code the evidence, then combine the codes. You assign each line of evidence a standardized code with a defined type and weight (population frequency here, computational prediction there, a functional study over here), and then a fixed set of rules rolls those codes up into one of five verdicts: Pathogenic, Likely Pathogenic, Variant of Uncertain Significance (VUS), Likely Benign, or Benign.
That two-step idea turned out to be the load-bearing structure we have built on ever since. Nearly every specific criterion from 2015 has looked at refining or replacing these criteria, but the structure is still there. Think of it as the operating system of variant classification: the apps keep getting updated, but they all still run on the same kernel.
Why the 2015 Framework Needed Modernizing
The original criteria were excellent for 2015. The problem is that they reflect the tools and assumptions of the year they were published. Several criteria leaned on in-silico predictors that have since been comprehensively outclassed. Others used population-frequency thresholds we now know to handle with more nuance. A couple proved redundant the moment richer data sources became routine.
The evolution of the guidelines builds on a couple of insights. First, some criteria acted like light switches, being on or off, even when the underlying evidence is more of a spectrum. A predicted loss-of-function variant isn’t always “very strong” evidence; sometimes it’s strong, sometimes only moderate, depending on where it lands in the gene. Second, the 2015 guidance was developed before the era of calibrated, AI-enhanced prediction, so it relied on methods such as “tool voting” with the best tools available at the time. We can now set statistically calibrated thresholds on a single effective tool.
ClinGen’s Sequence Variant Interpretation (SVI) working group took these on one at a time, publishing a stream of specifications that supersede the original criteria while leaving the code-and-combine logic untouched.

A Decade of ClinGen Specifications
Each major evidence type received its own renovation, overhauling how annotations and in silico predictions are applied. They change which variants move from VUS to actionable and which stay back. Here’s the tour.

PVS1, loss of function, from switch to dial. Abou Tayoun et al. (2018) replaced the single “very strong” loss-of-function call with a decision tree that follows the variant’s consequence and adds strength modifiers. A predicted null variant can now come in as PVS1_Strong, PVS1_Moderate, or even PVS1_Supporting, instead of always firing at full power whether or not it’s earned.
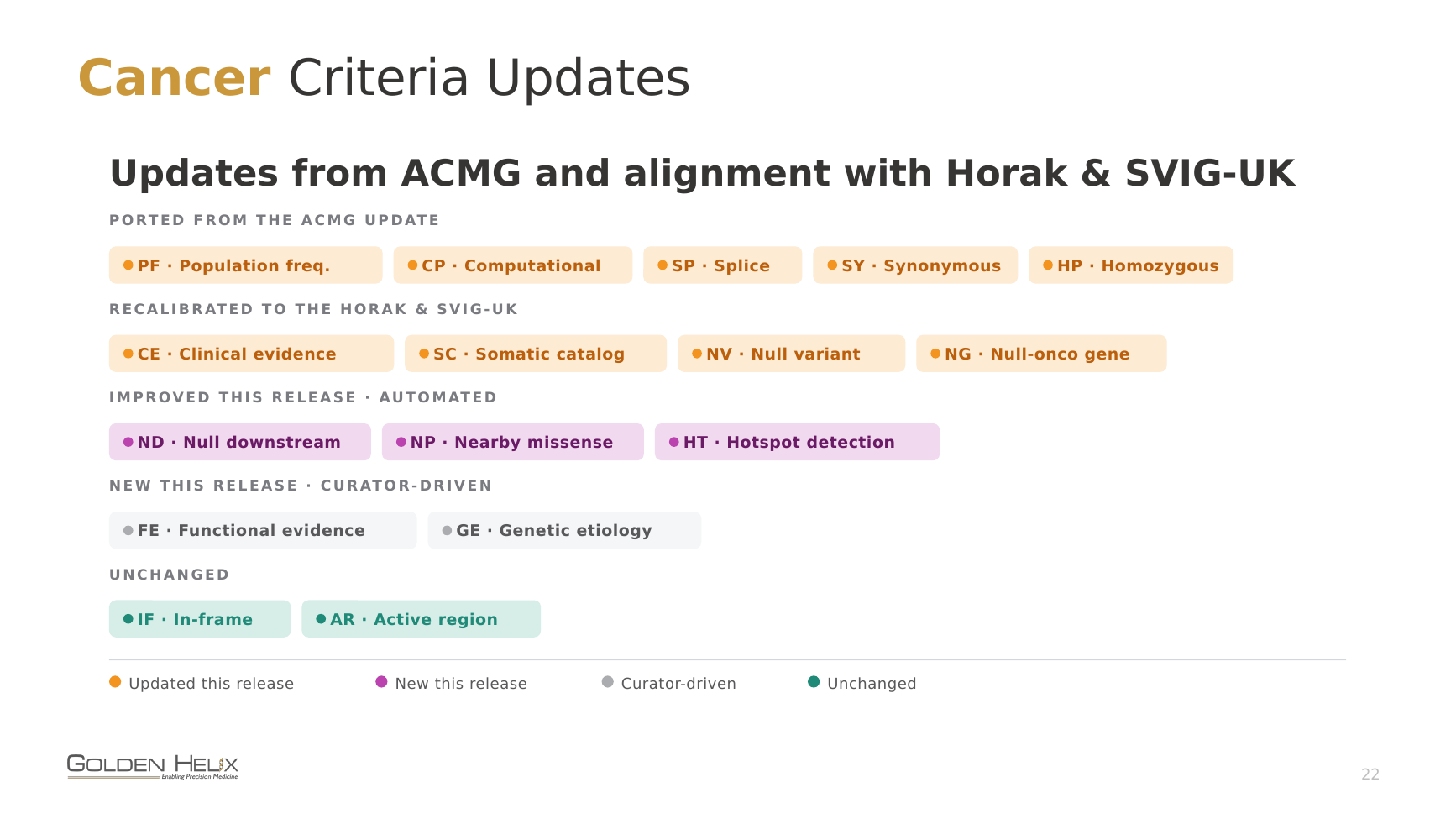
PP3 / BP4, missense, calibrated meta-predictors. Pejaver et al. (2022) retired the old “how many algorithms voted damaging?” approach in favor of calibrated thresholds on modern meta-predictors: REVEL, BayesDel, CADD. The payoff is range. These tools can reach up to Strong evidence, where the previous generation (SIFT, PolyPhen-2, raw conservation) topped out at supporting no matter how confident they were. That extra headroom is often exactly what lifts a variant out of VUS limbo on computational evidence alone.
PP3 / BP4, splicing, AI prediction. Walker et al. (2023) brought SpliceAI-class scores with calibrated cutoffs to replace the older multi-algorithm splice vote. The AI predictors are not only more confident; they can flag splicing effects well outside the canonical splice sites, the cryptic and novel splice variants the old tools simply couldn’t see.
BA1, BS1, PM2, population frequency, redone. ClinGen SVI moved the field from raw minor allele frequency to gnomAD with a filtering allele frequency (FAF), added an explicit BA1 exception list so a handful of common-but-disease-causing variants don’t get auto-benigned, and downgraded PM2 from Moderate to Supporting. FAF is, in plain terms, a more honest answer to “how common is this variant really?” It accounts for the uncertainty in the count rather than taking it at face value.
PP5, BP6, retired. These were the “a reputable source says so” codes, and they became redundant the moment we could pull ClinVar assertions in as evidence directly. ClinGen recommends dropping them, and the SVI papers also spell out how to stop accidentally counting the same evidence twice under two different codes.
VCEPs: Gene-Specific Rules That Win Ties
On top of the general specifications sits a layer of fine print: ClinGen Variant Curation Expert Panels (VCEPs) publish ACMG/AMP rules custom-tuned to a single gene or disease. A VCEP reweights the evidence around what’s actually known about that gene, and often redraws the pathogenic-side frequency cutoffs based on the disease’s real prevalence and penetrance. The rule of thumb is simple: where a VCEP exists, it wins. Its specifications take precedence over the generic defaults.
TP53 is the worked example. Rather than make analysts reconstruct the VCEP by hand for every variant, Golden Helix ships pre-computed criteria for each TP53 variant as a curated annotation track, aligned to the panel’s specification. The same pattern extends to genes like BRCA1/2, PTEN, MYH7, and a steadily growing list.
Cancer Variant Classification: Why Somatic Needs Its Own Framework
You might be thinking, “Why can’t the ACMG framework be used for any variant?” Well, there are good reasons we want a different classification scheme for somatic variants. A germline variant is judged on whether it might cause an inherited disease. A somatic variant in a tumor has to answer something else entirely: is it driving this cancer, and can we do anything about it? Those are different questions, so oncogenicity needs its own criteria, while still sharing infrastructure with ACMG wherever the underlying evidence is genuinely the same.
So, what makes a somatic variant oncogenic vs pathogenic? The mechanism is different, since gain-of-function oncogenes and loss-of-function tumor suppressors break in opposite directions. The frequency signal is inverted: what matters isn’t how rare a variant is in the population but how often it recurs across tumors. Recurrent mutated positions, the cancer hotspots, are some of the strongest impact predictors we have. And clinical actionability (does this tie to a therapy, a prognosis, a diagnosis?) is its own separate axis.

The framework still pulls a lot of shared evidence modeling from ACMG but uses a point-based scale in the spirit of Tavtigian et al., with a cancer-specific step that matches the disease mechanism, tumor suppressor versus oncogene. The original AMP/ASCO/CAP tiers (Li et al., 2017) defined four-tier clinical actionability, but not genomic variant oncogenicity. So in 2019, before any community consensus existed, we launched VSClinical AMP with the first real variant classification schema for cancer, defining an oncogenicity scale and code set that we presented to the VICC working group. That work fed into the 2022 ClinGen/CGC/VICC oncogenicity SOP (Horak et al.), which formalized point-based oncogenicity and benign criteria (the “O” and “SB” codes) validated on 94 variants across 10 genes. Most recently, the 2025 ACGS / SVIG-UK guidelines (Burghel, Mason et al.) published the first UK somatic guidelines covering both solid and haematologic tumors. It’s been rewarding to see a strategy we developed at Golden Helix adopted by international standards.
How VSClinical Automates Modernized Classification
To scale to the high-throughput demands of clinical genomics, we need an automated approach to applying these guidelines. VSClinical runs both the germline ACMG framework and somatic oncogenicity as guided, automated workflows, and has been updated to incorporate all the modernized evidence we have discussed.
On the germline side, the auto-classifier now routes splicing through CI-SpliceAI (which we showed to be functionally equivalent in sensitivity to SpliceAI), with new guidance thresholds and an awareness of nearby pathogenic splice variants. Missense calls are run on the BayesDel, REVEL, and CADD meta-predictors instead of the PolyPhen-2/SIFT/GERP++/PhyloP vote. Population frequency uses filter allele frequencies, per-gene ClinGen thresholds, plus the curated BA1 exception list. And the running point-based score sits right alongside the familiar criteria codes. The gene-specific frequencies and exceptions can be stored in editable, configurable assessment catalogs, rather than hard-coded, so your lab can review and adjust them to match your regional and disease expertise.
On the cancer side, the same in-silico evidence carries straight over (identical CI-SpliceAI and missense meta-predictors), the criteria are aligned to both the Horak 2022 SOP and the 2025 SVIG-UK guidelines in a single scoring strategy, and cancer-specific annotations like the Cancer Hotspots feed the score. Clinical evidence comes in-workflow through Genomenon CKB and our own Golden Helix CancerKB content. Because germline ACMG and somatic oncogenicity draw on one shared evidence engine, you can evaluate germline and somatic variants side by side in the same project, allowing detected germline variants and somatic variants to use their respective classification schemes.
Case Studies from the Demo
Let’s take a look at a couple of examples to see how these updates can make real changes to the way a variant would be classified.
Case 1: A VHL missense variant rescued from VUS
Take NM_000551.4(VHL):c.343C>T (p.H115Y), a missense variant in exon 2 of VHL. Under the old predictors, the simple amino-acid-substitution scorers, this one scored with benign computational evidence, which was enough to nudge it toward VUS. The modern meta-predictors flip the story. BayesDel calls it damaging (0.535) with a score high enough to provide Strong criteria evidence. Although we can review these various predictors, the guidelines state that a single predictor should be used consistently across all variants processed. While BayesDel is permissively licensed and can be used by commercial testing labs, REVEL can only be used in an academic context without a direct license. The missense paper shows them performing nearly identically, so our default in the software is BayesDel, but you can change the configuration to match your compliance preference.
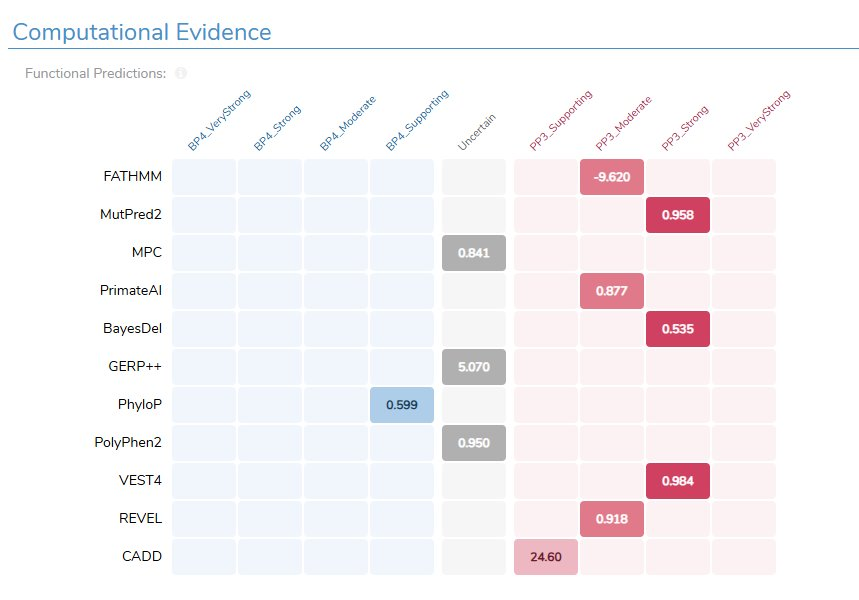
Case 2: A BRCA2 splice variant caught by CI-SpliceAI
Next, NM_000059.4(BRCA2):c.475+3A>G, sitting at a donor splice position. CI-SpliceAI predicts it disrupts the existing donor site, and the calibrated score is strong enough to land the variant just inside the Likely Pathogenic range. With the older, lower-resolution splice predictions, that evidence would have come up short, and a real splice-disrupting variant would have been filed under VUS. This is precisely the near-canonical, easy-to-miss effect the AI splice predictors were built to catch. (More on that annotation in Detect Cryptic Splicing Events with the New CI-SpliceAI Annotations in VarSeq.)
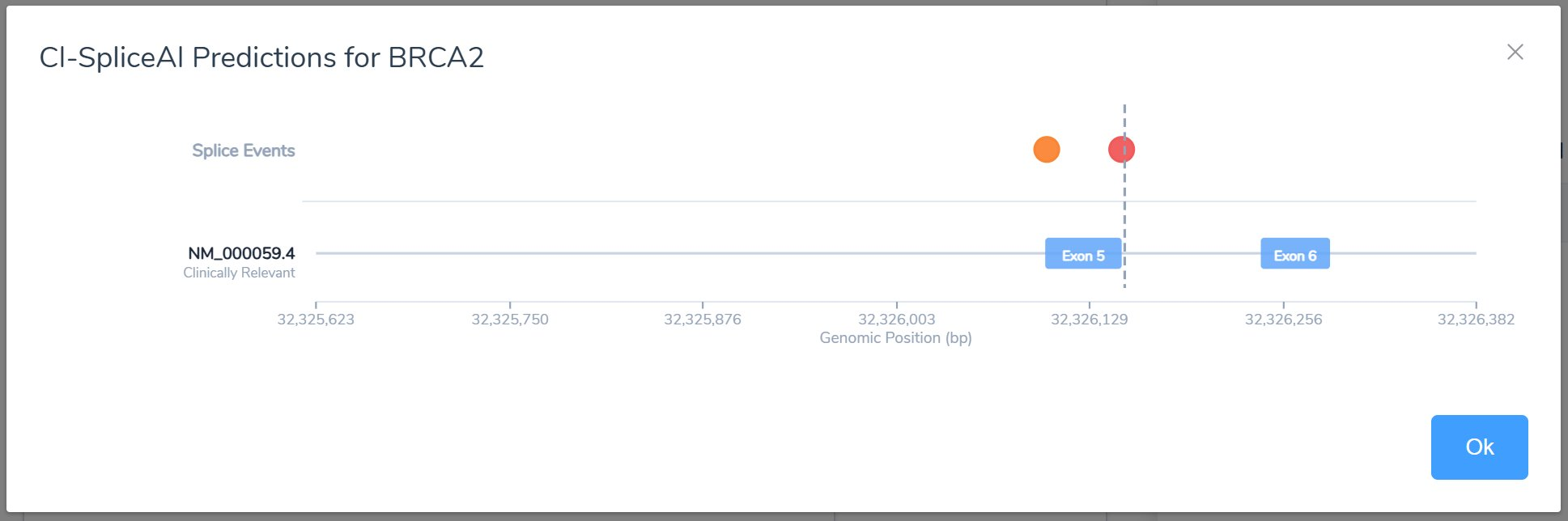
Case 3: Point-based scoring alongside the criteria codes
Along with a set of criteria, it’s helpful to look at the scoring process as a point-based system. In this example, the scored criteria (PS1 Strong, PM1 Moderate, PP2 Supporting) add up to +7 on the pathogenic side, landing the variant squarely in the Likely Pathogenic range (a score of 6 to 9). While the original criteria rules are still used, this point-based approach is helpful in adjudicating variants with conflicting evidence or on the edge of a classification boundary.
How Well Does Automated Classification Match the Experts?
A fair question to ask of any auto-classifier is: does it agree with a by-hand manual classification of variants? So, we benchmark both classifiers against published expert calls and review the classification concordance.
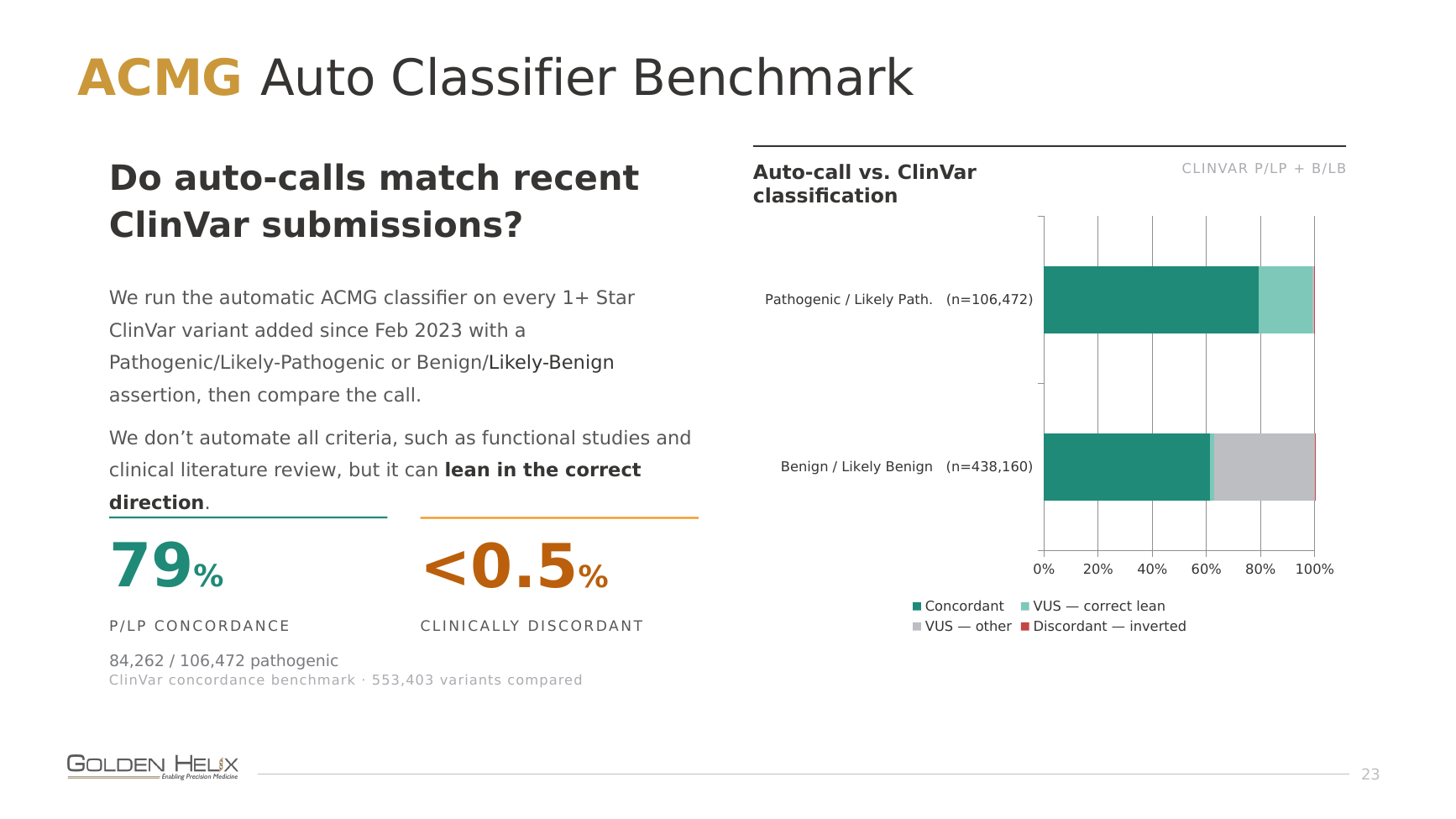
For germline, we ran the automatic ACMG classifier against every 1+ star ClinVar variant added since February 2023 that carried a Pathogenic/Likely-Pathogenic or Benign/Likely-Benign assertion (553,403 variants in all) and compared the calls. Note that the classifier was run with annotation data shipped strictly before this cutoff, so it was not influenced by the ClinVar submissions as clinical evidence. Strict concordance on the pathogenic side is 79%, and fewer than 0.5% of calls are clinically discordant (an inverted verdict). We deliberately don’t automate every criterion, since functional studies and literature review require human review, but even when the classifier can’t fully score a variant, it reliably leans in the right direction. We’re intentionally more conservative on the benign side, because we want to allow users to confidently filter out variants with these benign classifications.
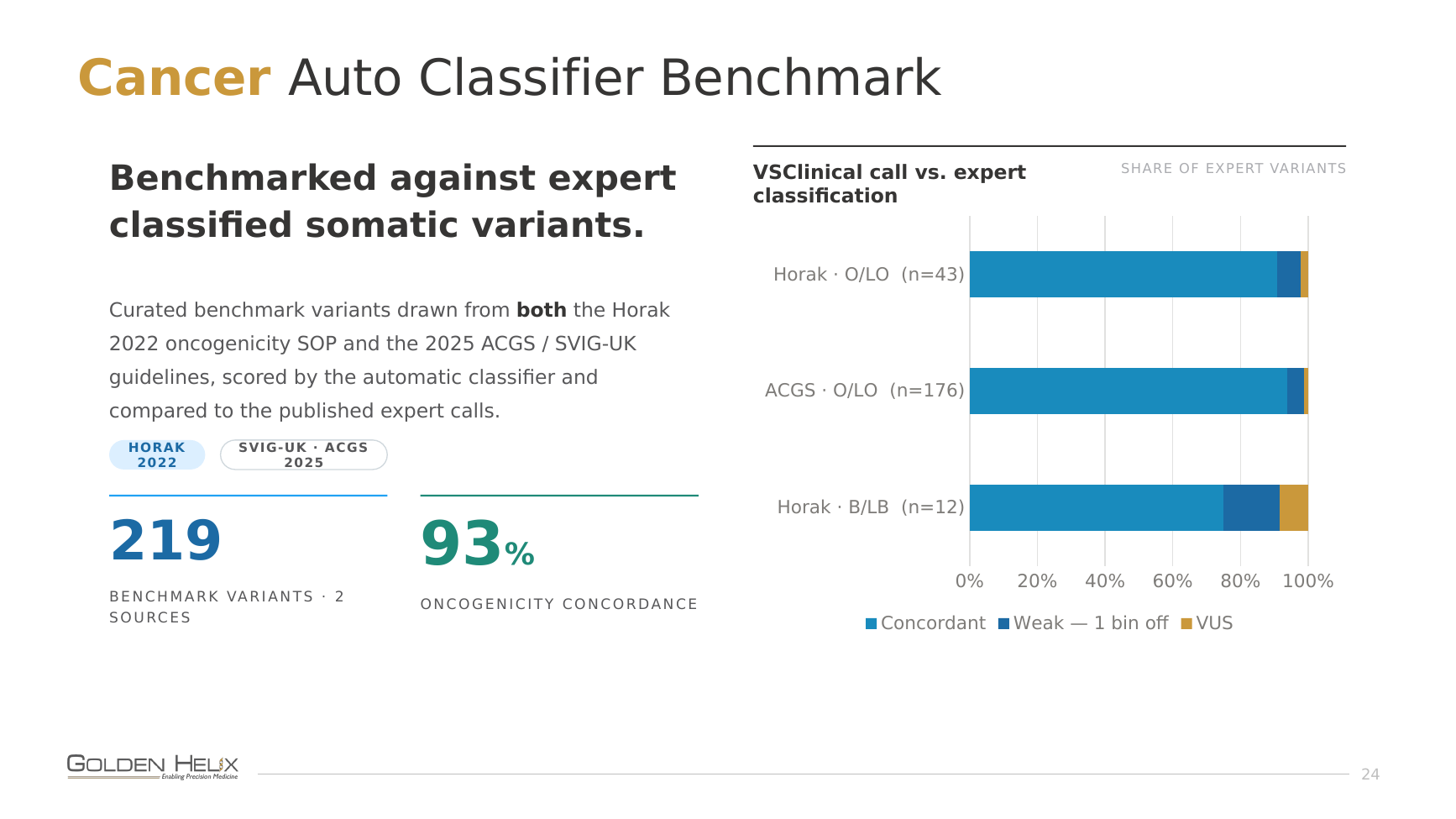
For cancer, we pulled curated benchmark variants from both the Horak 2022 oncogenicity SOP and the 2025 ACGS / SVIG-UK guidelines, scored them automatically, and checked them against the published expert calls. Across 219 benchmark variants from the two sources, oncogenicity concordance comes in at 93%.
Putting It Together
The 2015 ACMG/AMP framework gave clinical genomics a shared language, and its core move, code the evidence then combine the codes, has outlived nearly every specific criterion it shipped with. The decade since has been one long modernization: calibrated in-silico predictors, an AI splice model, a rebuilt population-frequency model, redundant codes retired, and per-gene VCEP rules layered on top. Cancer interpretation grew up next to it, answering its own question while sharing evidence wherever it can.
The payoff is we get fewer variants stranded in the VUS bin, leveraging modern algorithms and a more nuanced application of evidence. Keeping a pipeline current manually isn’t practical, so the best move most labs can make is to run on an analysis environment that implements the modernized specifications, allows reviewing previous variants with updated algorithms and annotations, and scores germline and somatic variants from one shared evidence engine.
That’s what VSClinical is for. To see the modernized ACMG and cancer classifiers in action, including the cases above, visit the VSClinical product page or explore the full VarSeq suite. And if you’d like to put your own variants through it, get in touch. I’m always happy to walk through real cases with a lab.
Frequently Asked Questions
What is ACMG variant classification?
ACMG variant classification is the standardized framework, introduced by Richards et al. in 2015, for interpreting DNA sequence variants. It defines a catalog of evidence criteria, each with an evidence type and strength, and rules for combining them into one of five categories: Pathogenic, Likely Pathogenic, Variant of Uncertain Significance (VUS), Likely Benign, or Benign.
How have the ACMG guidelines changed since 2015?
ClinGen’s Sequence Variant Interpretation working group and expert panels have published specifications that supersede most of the original criteria while keeping the code-and-combine logic. Key changes include strength modifiers for loss-of-function (PVS1), calibrated missense meta-predictors (REVEL, BayesDel, CADD) replacing tool voting, AI splice prediction (SpliceAI / CI-SpliceAI), a redone population-frequency model using gnomAD filtering allele frequency, the PM2 downgrade to Supporting, and the retirement of the PP5 and BP6 “reputable source” codes.
What is the difference between ACMG pathogenic classification and cancer oncogenicity classification?
ACMG classification asks whether a germline variant causes or contributes to inherited disease. Oncogenicity classification asks whether a somatic variant is driving a tumor and is clinically actionable. The cancer framework mirrors the point-based ACMG structure but uses cancer-specific evidence, recurrence and cancer hotspots rather than population rarity, and tumor-suppressor versus oncogene mechanism, formalized in the Horak et al. 2022 SOP and the 2025 SVIG-UK guidelines.
What is a VCEP and why does it matter?
A ClinGen Variant Curation Expert Panel (VCEP) publishes ACMG/AMP rules customized to a specific gene or disease, including gene-specific allele-frequency thresholds. Where a VCEP exists for a gene, and TP53 is the established example, its specifications take precedence over the generic defaults.
Can variant classification be automated?
Much of it can. An automated classifier can evaluate calibrated in-silico, population-frequency, and splice evidence and produce a point-based call, while leaving the criteria that need human judgment, such as functional studies and clinical literature review, to a curator. In Golden Helix benchmarking against ClinVar and expert-classified somatic variants, automated calls showed 79% strict P/LP concordance (germline) and 93% oncogenicity concordance (cancer), and leaned in the correct direction even when they couldn’t fully score a variant.



