
Aberrant splicing is one of the most consequential and least visible mechanisms driving human genetic disease. While variants disrupting the canonical GT/AG dinucleotides at exon–intron boundaries can be easily detected, some variants silently reshape splicing from deep within introns, far from the regions that targeted panels and whole exome sequencing (WES) were designed to interrogate. These deeply intronic variants are capable of activating cryptic splice sites and represent a significant source of missed diagnoses in rare disease analyses.
Detecting these variants requires two things working together: sequencing that reaches into the non-coding genome, and a splice prediction algorithm capable of evaluating each variant’s potential to alter splicing. In this post, we’ll explain how splicing works, why cryptic splice site activation is so difficult to detect, and how CI-SpliceAI can be used to annotate whole genome sequencing (WGS) data in VarSeq to identify likely pathogenic variants that would otherwise remain undetected.
Pre-mRNA Splicing and Cryptic Splice Sites
Pre-mRNA splicing is the process by which non-coding introns are removed from a gene’s initial transcript and the coding exons are joined into a mature mRNA. The spliceosome (the ribonucleoprotein complex that carries out this process) relies on conserved sequence signals at each exon–intron boundary: the GT dinucleotide at the 5′ donor site, the AG dinucleotide at the 3′ acceptor site, and surrounding regulatory elements. Splicing is further modulated by exonic and intronic splicing enhancers and silencers, making the regulatory landscape dense and highly sensitive to sequence variation.
Variants that directly disrupt the GT or AG dinucleotides are the easiest to detect and interpret as their disruption is almost always deleterious, and they are flagged routinely in any clinical pipeline. The more difficult problem is the large class of variants that affect splicing indirectly: those that weaken an existing splice site, alter an enhancer or silencer motif, or activate a latent cryptic splice site that the spliceosome would otherwise ignore. Cryptic splice site activation typically results in the insertion of a pseudoexon, a stretch of intronic sequence incorporated into the mature mRNA, leading to a frameshift, premature stop codon, or loss of protein function.
Deeply intronic variants that activate cryptic sites are particularly difficult to identify because they lie far outside the exon boundaries that targeted panels and whole exome sequencing (WES) are designed to capture. They are invisible to standard diagnostic sequencing and require both whole genome sequencing to reach the non-coding sequence and a splice prediction algorithm capable of evaluating the functional consequences of variants across the entire intronic landscape.
CI‑SpliceAI: An Open‑Source Alternative for Cryptic Splice Site Detection
SpliceAI transformed computational splice site analysis with the introduction of a novel approach to splice site detection based on a deep convolutional neural network trained on pre-mRNA sequences. Given a sequence window of up to 10,000 nucleotides centered on a variant, it predicts changes in donor and acceptor splice site probability, reporting four delta scores (acceptor gain/loss, donor gain/loss) that reflect the predicted magnitude of splicing disruption. High gain scores flag candidates for cryptic splice site activation, while high loss scores suggest disruption of existing sites. Its ability to capture long-range sequence context made it the leading tool for identifying deeply intronic splice variants.
Despite its impressive performance, SpliceAI’s restrictive licensing has created barriers for commercial clinical laboratories. CI-SpliceAI (Collapsed Isoform SpliceAI) addresses this by providing an open-source reimplementation of the SpliceAI architecture, retrained on a collapsed isoform transcript set derived from GENCODE annotations. Independent benchmarking, including a recent analysis by Golden Helix, demonstrates that CI-SpliceAI successfully replicates SpliceAI’s predictive performance with highly concordant delta scores across evaluation sets, making it a drop-in replacement without the problematic licensing constraints.
Golden Helix has made CI-SpliceAI annotation tracks available in VarSeq for manual review, with deeper integration coming soon. In the upcoming release, the VarSeq auto-classifier and VSClinical will automatically determine which ACMG criteria are relevant to each variant based on the updated recommendations from the ClinGen SVI Splicing Subgroup. VSClinical will also add splice impact visualizations that display predicted cryptic splice site locations relative to annotated exon boundaries, making it straightforward to assess the structural consequence of a predicted splicing change.
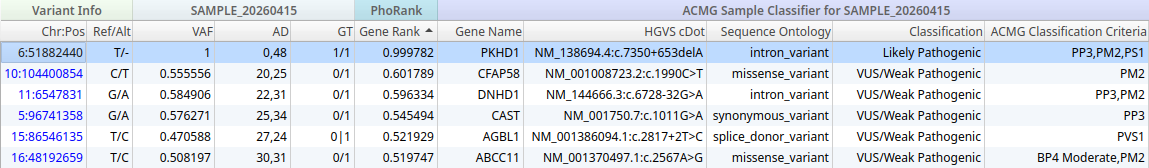
Identifying a Deeply Intronic PKHD1 Variant via WGS and CI-SpliceAI
To illustrate how CI-SpliceAI integrated with WGS can identify variants that standard workflows miss, consider the following hypothetical case, representative of the kinds of scenarios encountered in rare disease diagnostics.
A patient presents with renal insufficiency, systemic hypertension, and respiratory distress. The clinical picture is consistent with a disorder affecting renal tubular or collecting duct function, and the differential includes autosomal recessive polycystic kidney disease (ARPKD). Standard-of-care workup includes whole exome sequencing, which fails to identify any pathogenic or likely pathogenic variants in the coding exons of relevant disease genes. Given the strong clinical suspicion and the negative exome result, whole genome sequencing is ordered to survey non-coding and deeply intronic regions that WES does not cover.
Variant Prioritization in VarSeq
In VarSeq, the analysis is configured to filter for clinically relevant variants in genes associated with the patient’s phenotypes. Gene-level phenotype prioritization is applied using PhoRank, VarSeq’s phenotype-driven gene ranking algorithm, which scores genes for their degree of association with the patient’s HPO terms.
After filtering, a small set of six variants of interest remain. Among them is a deeply intronic variant in PKHD1, the gene encoding fibrocystin/polyductin, whose biallelic loss of function causes ARPKD. The variant (c.7350+653delA) sits well outside the nearest annotated exon boundary, beyond the region captured by WES, and would not have been visible in the exome dataset.
PKHD1 is ranked particularly highly by PhoRank, reflecting the strong phenotypic match between ARPKD and the patient’s presentation. The VarSeq auto-classifier has assigned a classification of Likely Pathogenic based on a combination of ACMG evidence criteria evaluated automatically based on the ClinGen SVI splicing recommendations.
Evidence Review in VSClinical
Opening the variant in VSClinical, we see that three ACMG/AMP evidence criteria are applied in support of pathogenicity:
- PM2: The variant is novel in both gnomAD and the 1000 Genomes Project. Absence from these large reference databases supports a pathogenic interpretation, as benign variants expected to be in the general population would be observed at detectable frequencies.
- PP3: CI-SpliceAI assigns a high delta score for acceptor site gain at this position, predicting that the variant activates a novel 3′ splice site deep within the intron. This score exceeds the ClinGen SVI working group threshold for PP3 evidence, indicating strong computational support for a damaging splicing effect.
- PS1: A different nucleotide change at the same intronic position, with an equivalent predicted splice site activation impact, has been previously classified as pathogenic in ClinVar. Under PS1 criteria, prior classification of a variant with the same functional consequence at the same genomic position provides strong evidence for pathogenicity.

The splice impact visualization in VSClinical displays the predicted novel acceptor site in the context of the surrounding PKHD1 exon structure. The visualization shows the predicted novel acceptor and donor sites along with their position relative to the canonical exon boundary.
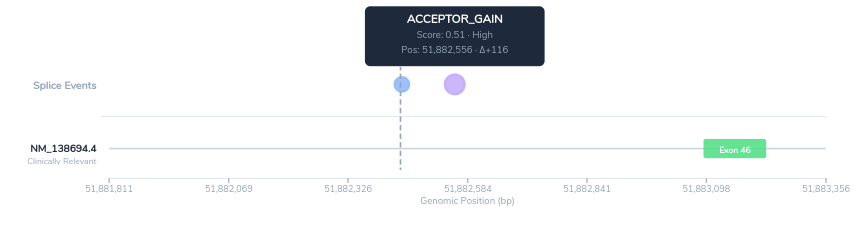
Taken together, the computational evidence, absence from population databases, prior clinical observations, and strong phenotype correlation support classification of this variant as Likely Pathogenic.
Conclusion
The case above illustrates a diagnostic pattern that is increasingly recognized in rare disease genomics: a patient with a compelling clinical presentation, a negative exome sequencing result, and an answer buried in the non-coding genome where standard exome/targeted panels cannot reach.
Because whole exome sequencing only captures the protein-coding exons and their immediate flanking splice sites, it misses the vast intronic landscape where cryptic splice sites can be activated by subtle sequence changes. Whole genome sequencing removes this constraint, but sequencing the non-coding genome is only useful if we have the analytical tools required to interpret what we find there.
CI-SpliceAI helps bridge that gap, applying deep learning to surface intronic variants that carry strong predicted splice impacts. Integrated into VarSeq’s filtering, ranking, and auto-classification workflow, it transforms a manual time-intensive search into a tractable clinical analysis.
The upcoming integration of CI-SpliceAI into the VarSeq auto-classifier and VSClinical represents a meaningful step toward the routine clinical detection of cryptic splice site variants. With VarSeq automatically handling the complex logic required to assess the ClinGen splicing recommendations, you can focus on the bigger picture, enabling faster sample processing and higher throughput, without sacrificing clinical sensitivity.
Frequently Asked Questions
What is a cryptic splice site?
A cryptic splice site is a sequence within a gene that resembles a canonical splice site but is not used under normal conditions, typically because it is outcompeted by the stronger, legitimate splice sites at the exon–intron boundaries. A mutation (often a single nucleotide change) can strengthen a cryptic site enough that the spliceosome begins to recognize it as a splice site, diverting splicing to this new location. Cryptic splice sites can be located in introns (leading to pseudoexon inclusion), in exons (leading to exon truncation), or at the boundaries of annotated exons.
What is a deeply intronic variant?
A deeply intronic variant is a DNA sequence change located within an intron, at a position far from the annotated exon–intron boundary, typically defined as more than 20–100 nucleotides from the nearest exon. Deeply intronic variants are outside the regions captured by most targeted gene panels and whole exome sequencing, which is why they represent a significant source of missed diagnoses. Despite their distance from exons, deeply intronic variants can cause disease by activating cryptic splice sites and inducing pseudoexon inclusion, as described above.
How does SpliceAI predict splice site changes?
SpliceAI uses a deep residual convolutional neural network trained on human pre-mRNA sequences and experimentally validated splice site annotations. Given a DNA sequence window (up to 10,000 nucleotides) centered on a variant, the model predicts the probability of splice donor and acceptor site usage at each position in the window, for both the reference and alternate alleles. The delta scores (the change in probability between reference and alternate) reflect how much the variant is expected to increase or decrease splicing activity at each nearby position. High delta scores for splice gain indicate likely cryptic splice site activation; high scores for splice loss suggest disruption of an existing site. SpliceAI’s architecture allows it to capture the long-range sequence context that influences splicing, enabling detection of deeply intronic activating variants that earlier tools consistently missed.
What is CI-SpliceAI, and how does it differ from SpliceAI?
CI-SpliceAI is an open-source reimplementation of the SpliceAI neural network architecture, retrained on a collapsed isoform transcript set derived from GENCODE annotations. Its core predictive model is functionally equivalent to SpliceAI and produces highly concordant delta scores across benchmarking datasets. The most important differences is its license, as CI-SpliceAI is freely available for commercial use. For clinical laboratories seeking to incorporate deep learning–based splice prediction into high-throughput WGS pipelines, CI-SpliceAI offers the performance of SpliceAI without the adoption barriers.



