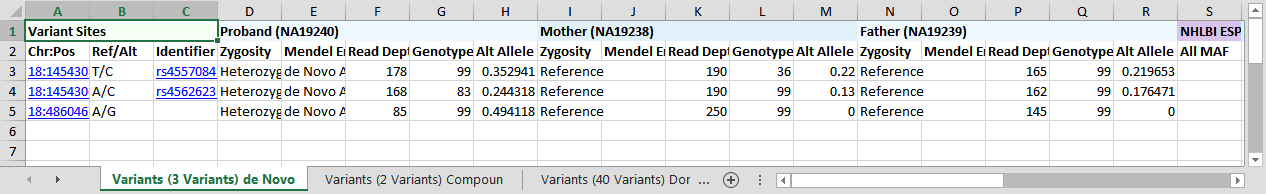
This project contains a Yoruban trio from the HapMap Project (Mother NA12938, Father NA12939, and Child NA12940). BAM files came from the 1000 Genomes Phase 3 Illumina Exome Alignment Project. We injected a de novo variant into the daughter's BAM file, then ran SAMtools for variant calling on all three family members simultaneously. The injected mutation is in the SMAD4 gene, associated with Myhre Syndrome.
Initial Workflow Summary
VCF data from chromosome 18 was imported using the Exome Trio Template, with the pedigree structure set during import. The result: 734 total imported variants. Additional annotation was applied from dbNSFP Functional Predictions 3.0 (GHI) and ExAC Variant Frequencies 0.3 (BROAD).
Because the proband is female and the project only contains chromosome 18 data, we disabled the X-linked filter chain.
Investigating Filter Chains
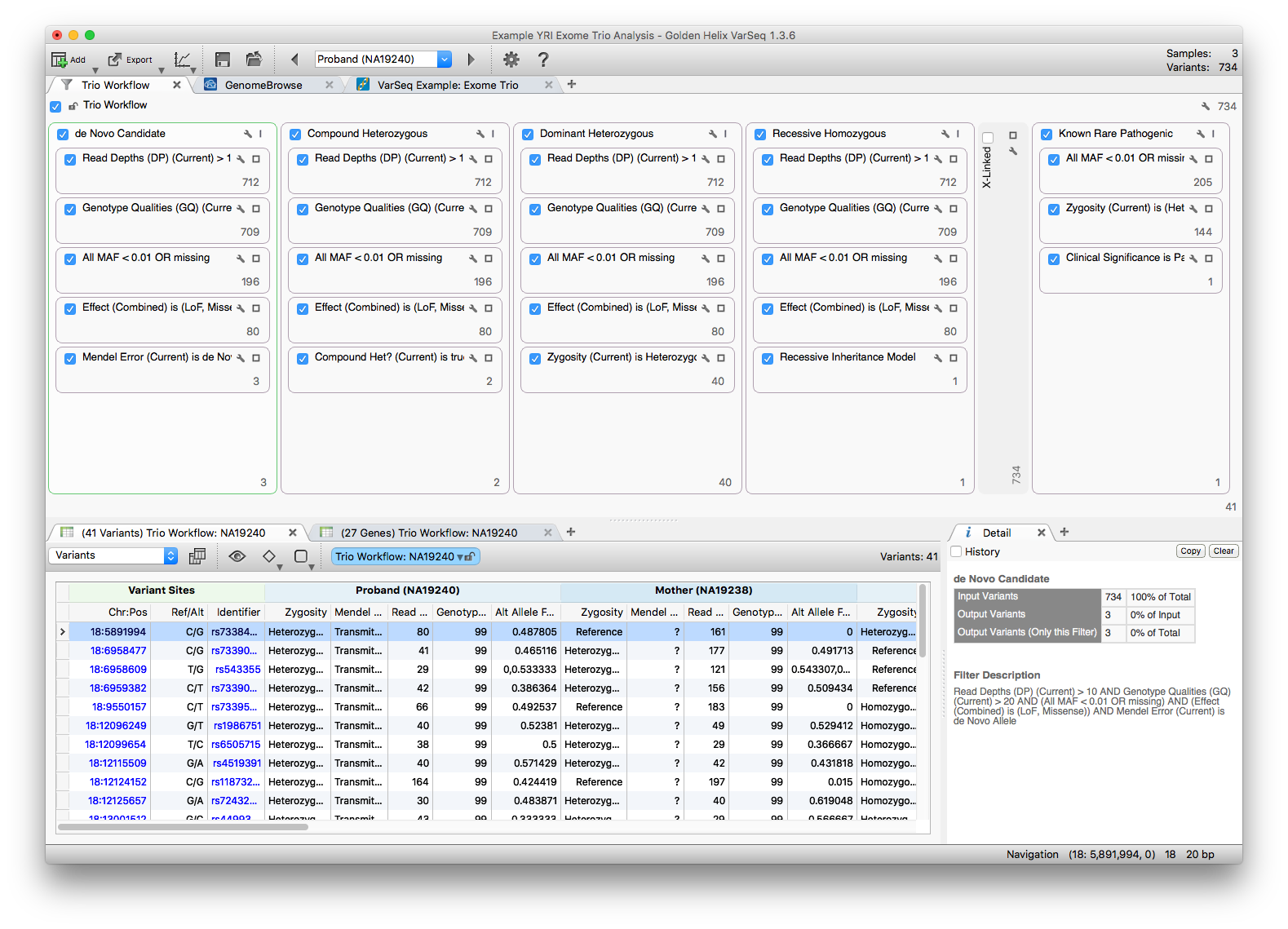
Five filter chains run in parallel against the same 734 variants, each looking for a different inheritance pattern. We will examine the de Novo Candidate, Compound Heterozygous, and Dominant Heterozygous chains.
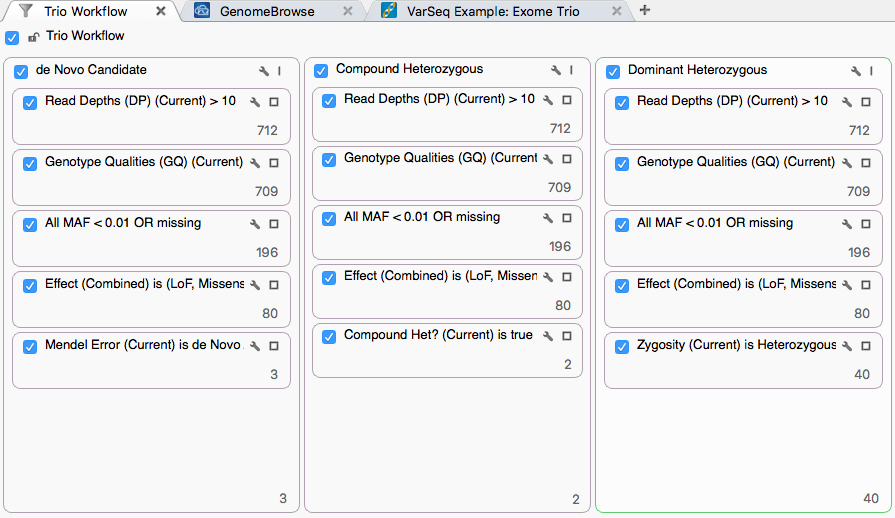
Each chain shares a basic-quality stage, then applies an inheritance-specific filter. The shared stage uses:
- Proband Read Depth (DP) > 10
- Proband Genotype Quality (GQ) > 20
- NHLBI All MAF < 0.01 or missing
- Variants classified as Loss of Function or Missense per RefSeq
These shared filters reduce the set to 80 variants, which then feed each inheritance-specific filter.
De Novo Candidate
Click the 3 at the bottom of the de Novo container to view the three variants that survive this chain. The specific filter card uses output from the Mendel Error algorithm (available under Add > Computed Data).
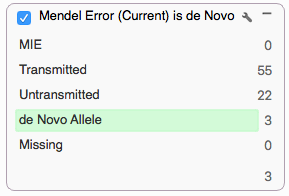
A de novo classification requires the proband to be heterozygous and both parents to be homozygous reference or missing. To validate findings, the BAM files were loaded into the GenomeBrowse tab alongside the variant table. Click through table rows to zoom GenomeBrowse to the matching position.
For the first variant, 18:14513039 T/C (rs45570841), the BAM files for both parents show reads supporting the alternate allele.
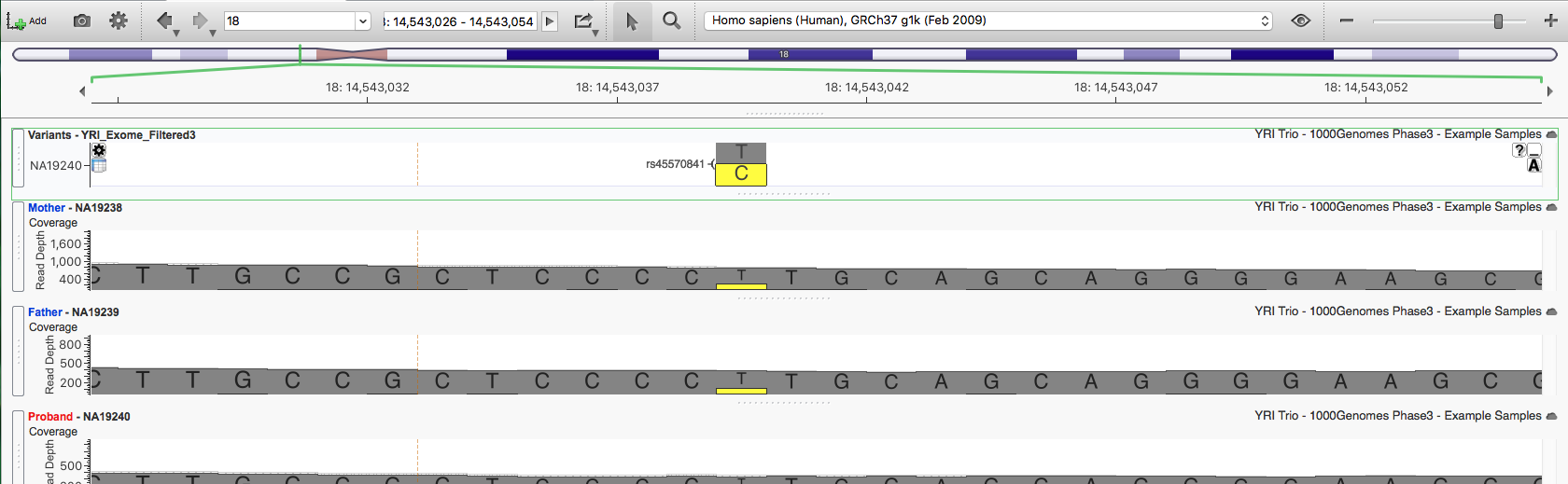
The variant caller called both parents reference despite roughly 20% alternate-allele reads in each. We can dismiss this candidate as a false positive. The same is true for the second variant, 18:14543063 A/C (rs45626231). Scrolling the visible annotation columns confirms neither variant is in ClinVar, and all six common dbNSFP algorithms predict them as Tolerated.

For the third variant, 18:48604676, neither parent has any reads supporting the alternate allele, making it a strong de novo candidate.
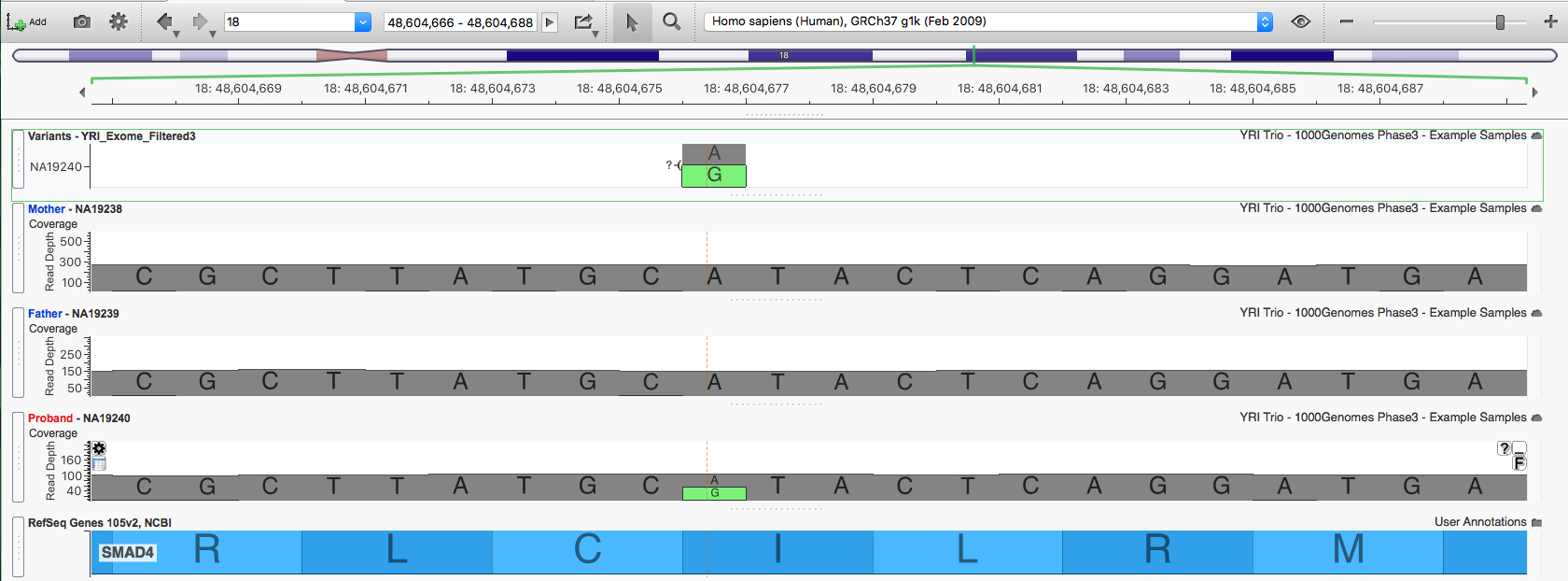
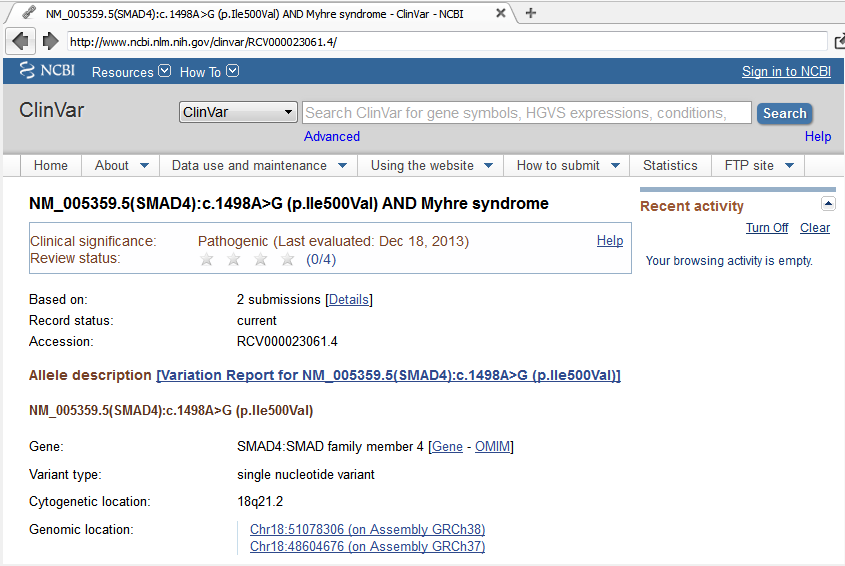
The Detail pane shows this is a missense variant classified by ClinVar as Pathogenic for both Myhre Syndrome and a Hereditary cancer-predisposing syndrome.
Click the ClinVar Accession identifier (RCV000023061.4) in the details pane to open the ClinVar record in the VarSeq browser view.
Compound Heterozygous
Click the 2 at the bottom of the Compound Heterozygous container to view the two variants that pass this chain. The specific filter uses output from the Compound Heterozygous Detection on Trio algorithm.

A compound heterozygous polymorphism means a child inherited two different heterozygous polymorphisms in the same gene, one from each parent. Both copies of the gene are potentially affected, and the variants should be non-synonymous.
This trio has one identified compound heterozygous region in the TPGS2 gene. Click the (1 Genes) Compound Heterozygous: NA19240 table to see the two variants that make up the region. Both are classified as Transmitted by the Mendel Error algorithm, indicating inheritance.
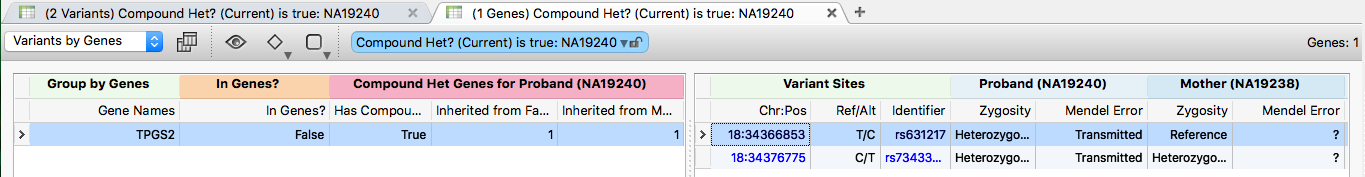
Click a variant in this region to zoom GenomeBrowse to the location, then double-click the TPGS2 gene in the RefSeq annotation track to zoom out to the full gene region (chr18:34,359,987-34,409,179), including both variants of interest (rs631217 and rs73433599).
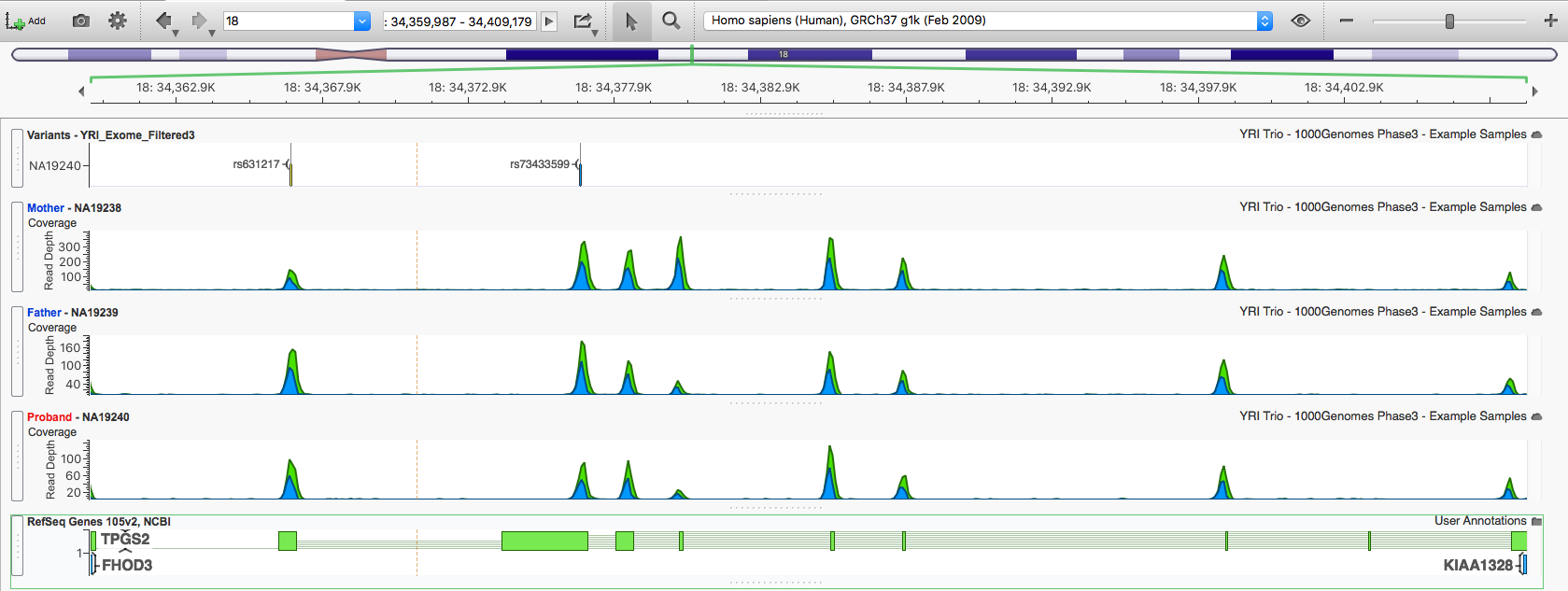
Scrolling the annotations shows neither variant is in ClinVar, but rs73433599 is classified as Damaging by 3 of the 5 prediction algorithms in dbNSFP.
Dominant Heterozygous
For a more compact view of multiple parallel filter chains, minimize the chains you are not currently working with.
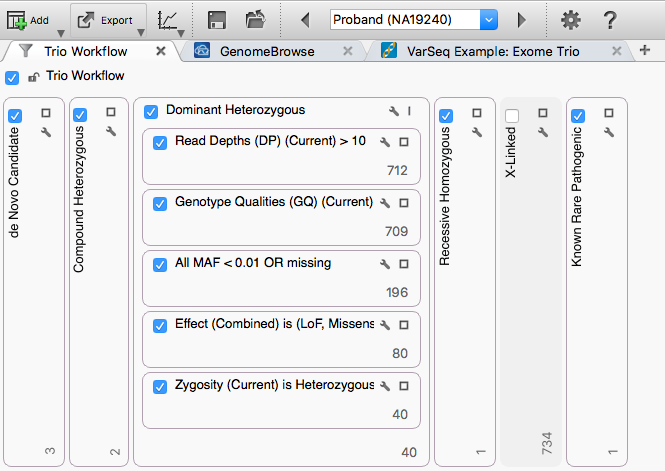
Click the 40 at the bottom of the Dominant Heterozygous container to show the 40 variants that pass this chain. The specific filter uses output from the Genotype Zygosity algorithm.

Several options narrow the list further: keep only variants predicted as Damaging by at least 3 of the 5 dbNSFP algorithms, drop de novo variants flagged by the Mendel Error algorithm, or annotate with additional sources and filter for rare variants (for example, by ExAC frequency).
Exporting Data
The Excel export supports multi-tab export of variant tables in one pass, so the results from all three filter chains can ship in a single file.
Reset the variant table view to the de Novo filter chain by clicking the 3 at the bottom of the chain, then delete the (27 Genes) Trio Workflow: NA19240. Lock the view by clicking the lock icon on the blue filter chain button.
Click the + sign to add a new table view.
When the new table becomes active, click the 2 at the bottom of the Compound Heterozygous filter chain.
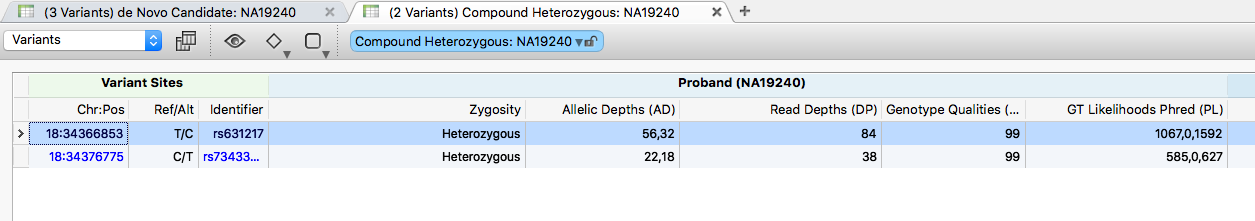
Repeat to add the results from the Dominant Heterozygous filter chain.
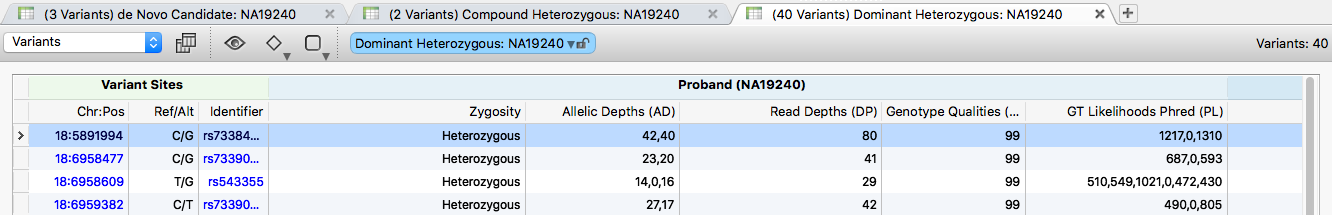
To export, go to Export > XLSX File and leave all tabs selected.
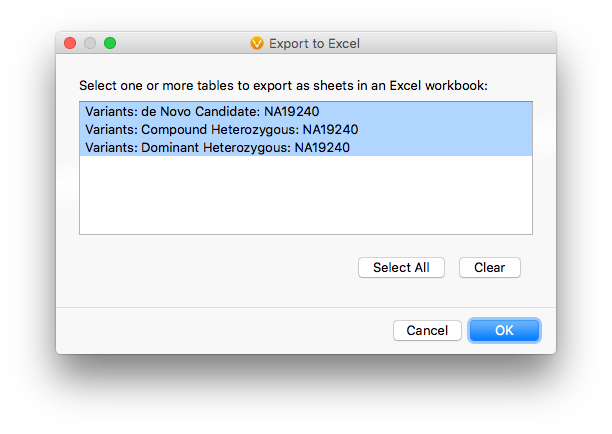
Leave the default visible-field selection and click Export. When the export finishes, open the single file containing tabs for all three chains.