It may be possible to say that annotating a variant correctly and accurately against gene transcripts is the most important job of a variant annotation and interpretation tool. We take it very seriously at Golden Helix as we support VarSeq and its use by our customers in both research and clinical contexts.
It has been a source of frustration that the default gene track for the commonly used GRCh37 genome reference is quite out of date from the current NCBI RefSeq database. In fact, it was created in 2013 as part of the 105 Annotation Release!
Although new versions have come out on the latest GRCh38 genome reference, their policy has been to annotate “backward” to the previous, but still widely used 37 build.
Recently, responding to the suggestions and pressures from myself and other in the genomics community, NCBI provided a “interim” release that aligned the latest RefSeq gene database back to GRCh37.
We have updated our default gene track based on this much-improved resource, as well as added some additional fields to our transcript annotation algorithm.
Genes Models Improve, and So Should Our Annotations
Although we often think of genes as being very concrete and permeant parts of the genome, the reality is that our understanding of which transcripts encode viable protein products and which of the many computationally derived transcripts are active and detected in organisms is a constantly evolving knowledge base.
Even commonly interpreted genes in human genetics can have their potential transcript list expanded. For example, PMS2 is often included in hereditary cancer panels, and in the latest RefSeq release grew from one “NM” transcript to fourteen!
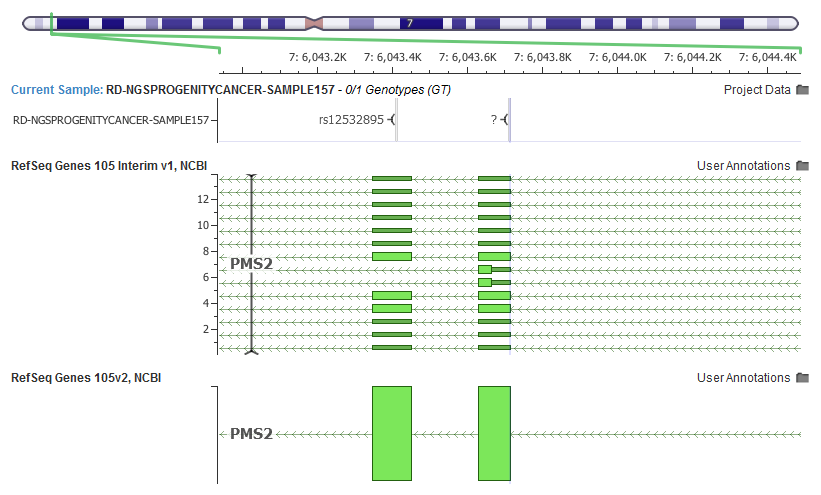
Along with the updated transcript definitions from RefSeq, we also pulled in the latest LRG ID mappings that provides us with the primary metric in which the “Clinically Relevant” transcript is selected using our ClinVar derived heuristics.
Not Just Newer, But Better
Finally, as I enumerated in the 2014 blog post where we carefully updated our default track to be based on NCBI RefSeq, there are genes in the GRCh37 genome assembly that do map cleanly due to errors in the reference genome and the choice of gene annotations sources also is a choice of the way these errors are handled.
While ultimately I felt NCBI provided the best overall transcript alignments of their own RefSeq database, there were edge cases where their handling of these genome reference errors resulted in more difficulty in the downstream annotation and interpretation process.
This is exemplified in the SRD5A2 gene, which is often included and interpreted in hereditary gene panels. In GRCh37, it is missing a single letter in the very first exon in the human reference that is part of the canonical transcript. The v105 RefSeq didn’t account for this in its alignment resulting in a reading “frame-shift” in our in-silico transcript construction and ultimately problematic variant annotation for the rest of the gene.
While this genomic reference error is removed in GRCh38, some of our clinical lab customers use this gene in their panels and had to do manual inspection and work-around for interpretation of variants with the GRCh37 assembly and v105 transcripts.
With this latest “interim” release, the NCBI alignment algorithm does a trick used by the UCSC and Ensembl alignment algorithms and introduces a “mini-intron” to account for the missing letter and keep the rest of the gene “in-frame”.
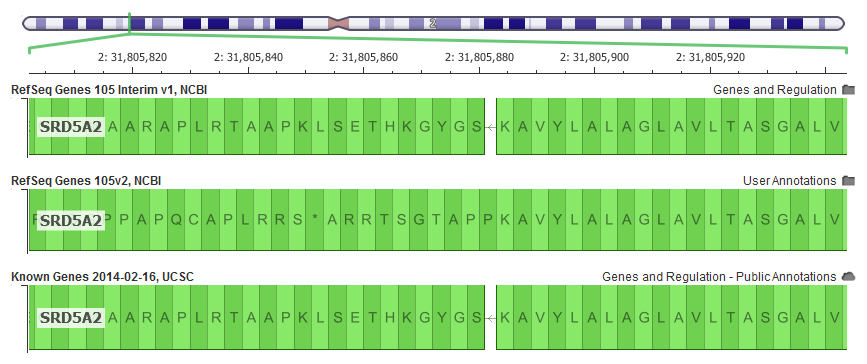
The latest RefSeq Gene annotations are on top, and are an improvement on the previous default 105v2 alignments that where quickly out of frame. Notice the “*” stop codon 10 codons downstream (to the left of this reverse strand gene)!
New Fields to Support Interpretation
As VarSeq has been adopted by more clinical labs that use it to power their variant science, we have received several requests to expand our gene annotations outputs to provide more details for certain classes of variants.
With the release of VarSeq 1.4.4, we incorporated this feedback into the algorithm with the addition of a number of fields and improvement of existing fields.
These include:
- New fields in the Transcript Interactions source that provide the Reference and Alternate Amino Acid for each transcript in the interactions table.
- Add a field to capture the number of total exons that are present in each transcript. Along with the existing field for which exon a variant is in, this allows you to report a variant is in exon 9 of 20 for a given gene transcript.
- Intronic variants now have their nearest exon reported in the Exon Number field, and a new field 5’ Exon Number can be used to determine which two exons the intronic variant is between. This allows more descriptive reporting of intronic variants.
A Release with a Hundred Contributors
Ultimately, VarSeq matures as a function of its fantastic adoption in the clinical genomics and research market.
If you read through our massive release notes for VarSeq 1.4.4, you can get a sense of our process of incorporating the hundreds of comments and insights from our ongoing engagement with customers back into making our products better.
I want to really commend NCBI for listening to their customers, and putting the resources into building a new and much improved RefSeq gene annotation as well.