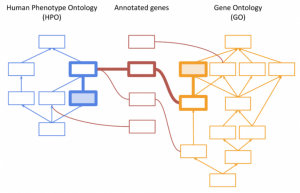
Integrating Genomenon Mastermind Curated Variants in VarSeq and VSClinical

When performing variant classification using VarSeq’s ACMG classifier, a large number of variants inevitably fall into the category of variants of uncertain significance (VUS). This is because much of the evidence required for confident classification cannot be programmatically evaluated. Take PS3, for example, the criterion for functional evidence. This criterion…
Read more →