Considering Data Quality: From Sequencing to Reporting

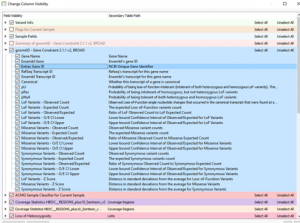
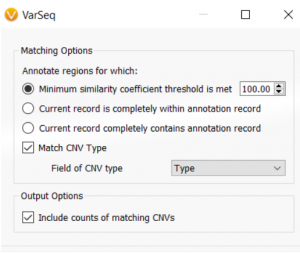
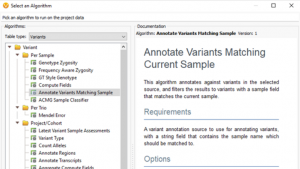
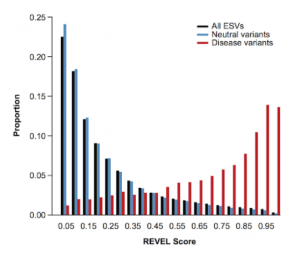
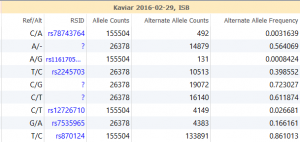
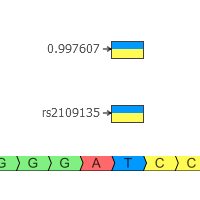
When quality metrics generated during secondary analysis are not incorporated into interpretation and reporting, variants are reported without the important context that could affect confidence in the call. The Golden Helix software suite has been designed to help you bring in quality metrics from secondary analysis through to interpretation and…
Read more →