In the first part of this series, we covered how VarSeq supports customizing ACMG guidelines in VSClinical to streamline the clinical analysis workflow. VSClinical’s various customization parameters within the ACMG Guidelines workflow includes the choice of how the internal knowledge base of previous variant interpretations are stored and what considerations go into this choice. Customizing ACMG guidelines in VSClinical includes decisions around interpretation thresholds, population frequency cutoffs, and preferred data sources.
Greater Than Expected for the Disorder
The ACMG guidelines for the classification of sequence variants often phrase the criteria by which variants are scored in terms that require the expert in the genomic and disease context to make the concrete rule. For example, the rule BS1 should be scored if a variant’s allele frequency in non-diseased populations is “greater than expected for disorder”. While the paper provides some examples of carrier frequency for some recessive disorders, no single threshold would cover every testing scenario.
VSClinical provides a recommendation engine that evaluates as many ACMG criteria as can be automated and highlights the suggested scored criteria and provides the reasons and evidence for the recommendation. To provide a recommendation for the three population-frequency focused criteria BA1, BS1 and PM2, a different set of thresholds were chosen for Recessive versus Dominant genes. The defaults for these values were carefully chosen from the literature, expert consultation and empirically testing of classified variants with consensus classifications. But these thresholds can be changed to suit the genetic test being performed by a lab when configuring the project.
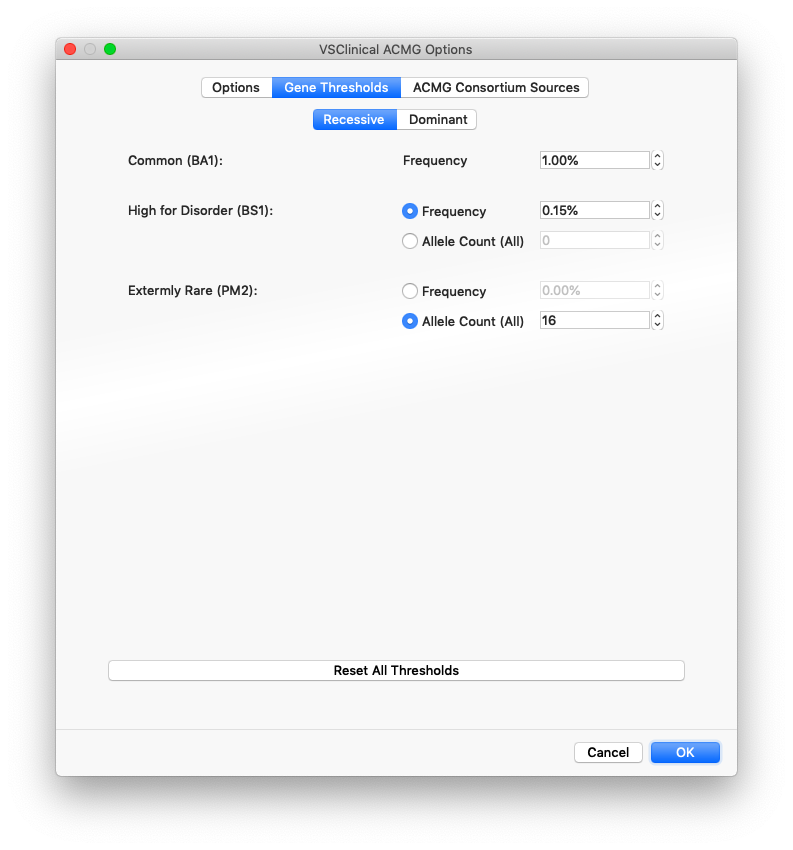
The options when choosing the thresholds for BS1 and PM2 include using a specific frequency value based on the maximum sub-population allele frequency across the major population groups in gnomAD exomes and the 1000 Genomes Project or from using a specific Allele Count (AC) value across populations (the entire catalog).

Established Pathogenic Variants
Another source of customization a lab may explore is amending ClinVar with its own source of established Pathogenic Variants. Of course, the variants previously classified by the laboratory will be included in future recommendations when looking up pathogenic variants in the same position, amino acid, or hot-spot region for the purposes of establishing a recommendation for various ACMG scoring criteria. But beyond the internal knowledge base and ClinVar, other “consortium” sources can be consulted as a source of clinically classified variants.
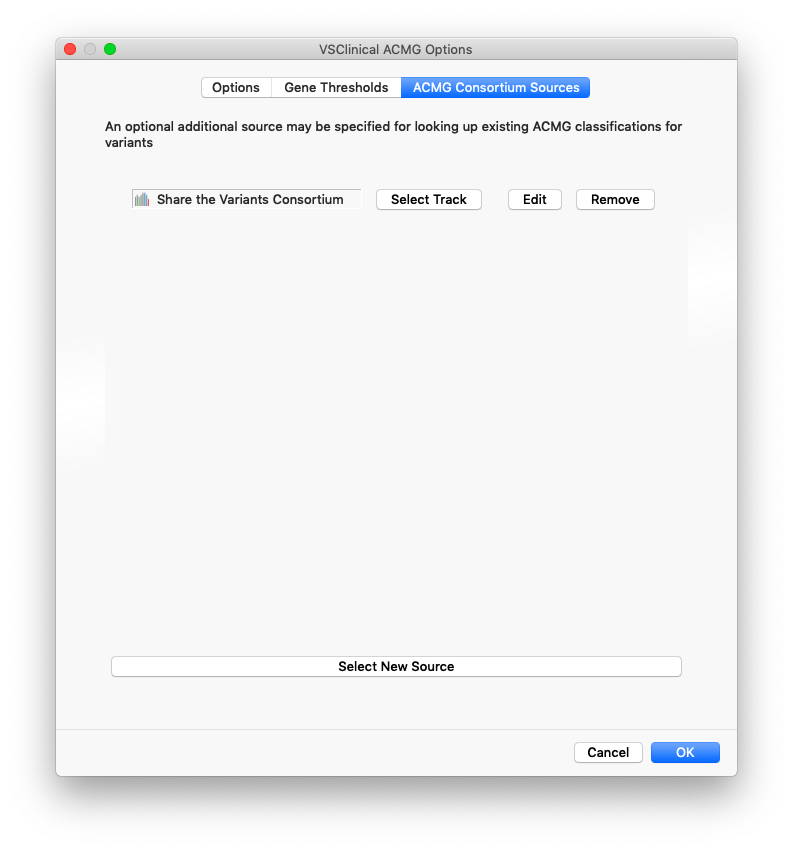
In the ACMG Consortium Sources tab the VSClinical configuration dialog, these additional sources can be added. There are four fields that can be mapped and used by VSClinical to query these sources for relevant variants:
- Classification: This field should have values from the ACMG Classification tiers of Benign to Pathogenic and will be used to determine if a variant or nearby variant was previously classified as Benign or Pathogenic.
- Rating (Optional): If a rating is provided, it will be used to ignore variants with a “(0 stars)” rating.
- Condition (Optional): An optional condition will be used to provide the user context of what condition the classification was interpreted.
- Date (Optional): In a parable string or UTC UNIX timestamp form, this will be presented to the user as the date the classification was made. An example would be “2019-07-21”.

A consortium source will also add to the classified variants in the table of the Gene Region and Mutation Profile section. This table helps inform the tricky “Located in mutated hotspot” PM1 criteria.
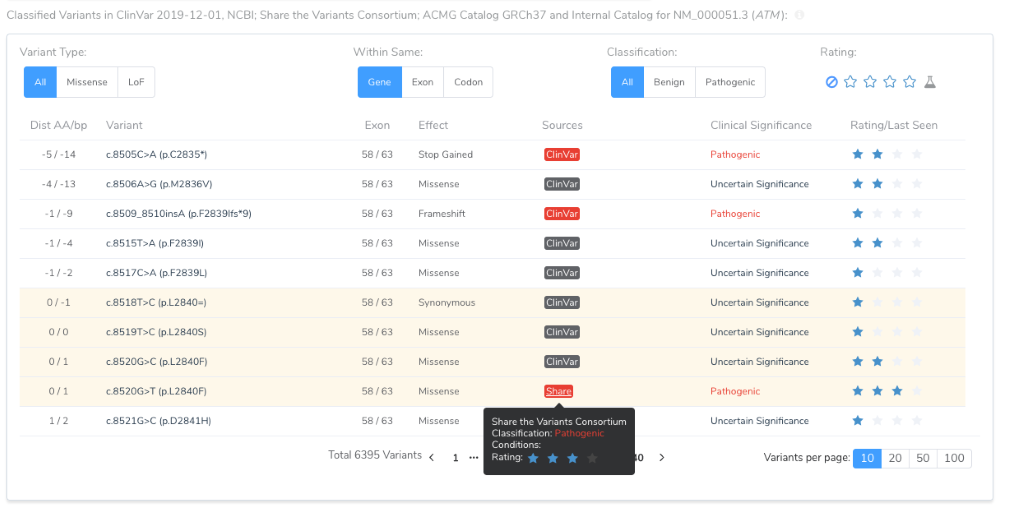
For a given variant, if multiple sources provide a classification, the following precedent is used to determine pathogenicity:
- Lab Catalog
- Consortium 1
- Consortium 2…N
- ClinVar 1+ Star Clinical Significance
So once a variant is classified by your lab, regardless if it is classified as Pathogenic, Benign or even as a Variant of Uncertain Significance, that classification will be used for future variant evaluations. At the same time, VSClinical will make it clear that other classifications exist.
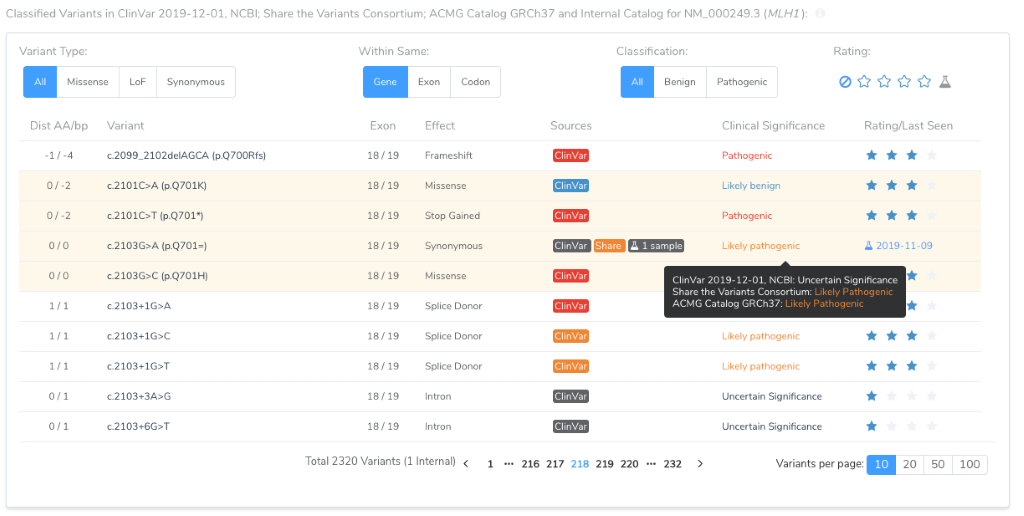
VSClinical is designed to be broadly applicable to most genetic testing scenarios out of the box. We did extensive testing of the automatic scoring recommendation against previously classified variants in ClinVar and other sources and configured defaults and heuristics that provide excellent performance. But for specific testing scenarios and test designs, these defaults can be customized and expanded with lab-specific expertise. In the next installment of this series, we will go into per-gene level preferences and how VSClinical helps keep track of the changing landscape of annotations, transcript selection, and inheritance models.



