I remember visiting a patient in the NICU amongst the incubators, some glowing blue like tiny tanning beds to treat jaundice, all containing tiny humans – many smaller than a loaf of bread. Infants get admitted into the neonatal intensive care unit or NICU for many reasons ranging from elevated bilirubin, hypoglycemia, sepsis, and respiratory distress (RDS). Many are eventually diagnosed with an underlying genetic condition. Ideally, any genetic issues are identified by non-invasive prenatal testing (NIPT) and, if needed, Amniocentesis in the second trimester. However, these tests are typically limited in scope and rely on biochemical assays, karyotyping, and DNA probes to test for chromosomal disorders and microdeletion syndromes.
Each year ~6% of children born worldwide have a genetic birth defect and contribute significantly to morbidity and mortality. Half of these defects have a known origin, 70% of which can be treated or prevented entirely in developed countries (Christianson et al., 2006). That leaves the other half – ~3% of infants born with an unidentified genetic disease, underscoring the need for robust rare disease analysis capabilities. In either case, traditional diagnostic approaches are long, invasive, and costly. With time working against you, NICU providers are increasingly turning to genomics, achieving up to 60% diagnostic yields (Gubbels et al. 2020). But the realization of genomic medicine in clinical practice rests on efficient and accurate genomic interpretation. Knowing that various gene panels could miss the genetic culprit, many providers opt for whole exome sequencing or whole genome sequencing. While there’s a good chance of capturing your variant of interest with these methods, the crux is identifying it out of hundreds of thousands of possibilities. Ultimately, the effectiveness of genomic medicine hinges on efficient and accurate data interpretation. Here’s where those little loaves of bread come in. Every neonate is in the NICU for a reason, and those reasons, that phenotypic information, is statistically tractable.
Take this example. You have a 3-week-old infant, Lauren, whose parents have brought her in after observing seizure-like behavior. Lauren had previously been in the NICU due to hydrocephalus and subsequently received a ventricular peritoneal shunt to reduce the fluid around her brain. Dermatological redness and a small verrucous blister were also noted in her chart, and you notice ridging in her fingernails. With little else to go on, you opt for a genomic approach. Upon opening your results in a VCF file, you’re confronted with over a hundred thousand variants. You’re able to use quality metrics, specifically read depth and Phred score, to get rid of low-quality variants and allele frequency from population catalogs like gnomAD and 1kG Phase3 to eliminate common variants. Still, you’re left with over 11,000 possibilities. But that number can start to change when you incorporate phenotype.

Aptly named, algorithms like PhoRank rank genes based on their relevance to user-specified phenotypes as defined by the GO and HPO biomedical ontologies for both SNPs and CNVs.
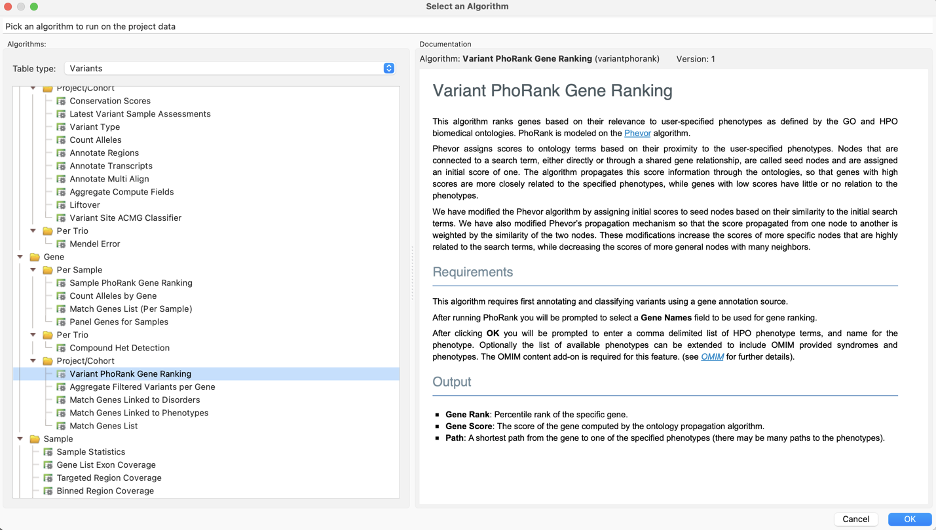
We can enter Lauren’s phenotypes and get gene scores to add to our filter chain, helping prioritize variants.
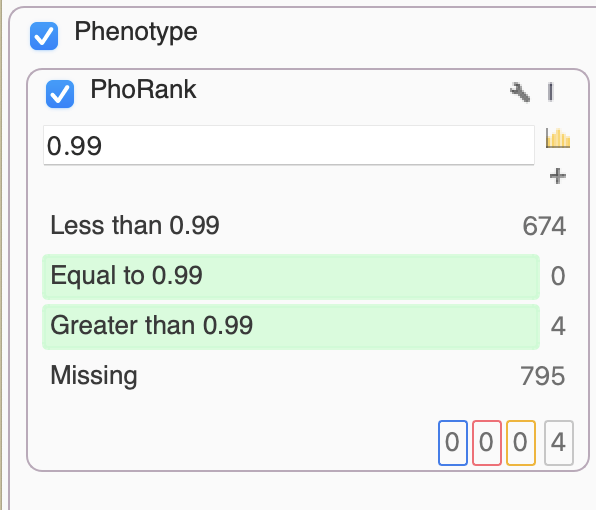
Alternatively, a similar algorithm for this prioritization is the Match Genes Linked to Phenotypes. This comes in handy when we want to avoid the “rank” specificity output of PhoRank, and instead consider any associated genes in their entirety based on phenotypic terms. In this scenario, we only needed PhoRank to whittle things down to just 4 variants, one of which stood out as a G>C of IKBKG, resulting in loss of function.
This mutation can be responsible for Incontinentia pigmenti, an X-linked dominant multisystem neurocutaneous disease that can be particularly difficult to diagnose in the neonatal period often mistaken for a herpes simplex infection (Rodrigues et al. 2011). With the power of phenotypic and genomic data combined, Lauren wasn’t subjected to unnecessary antivirals, and her immediate and long-term care can be tailored to her disease.
In the end, mechanisms that leverage phenotype like PhoRank will save individual lives like Lauren and inform the broader clinical management of patients. Statistically tractable phenotypic information in genomic contexts will lead to improved mechanisms for capturing and sharing this data. As a result, these contributions will increase genetic analysis capabilities for rare diseases by enabling the identification of multiple unrelated patients with similar phenotypes – a crucial step toward ending diagnostic odysseys. If you have any questions regarding phenotype info, please reach out to us at [email protected].
Christianson, A.C., Howson, C.P., Modell, B. (2006) The March of Dimes global report on birth defects: the hidden toll of dying and disabled children. White Plains (New York): March of Dimes.
Gubbels, C.S., VanNoy, G.E., Madden, J.A., et al. (2020) Prospective, phenotype-driven selection of critically ill neonates for rapid exome sequencing is associated with high diagnostic yield. Genet Med 22, 736–744.
Rodrigues, V., Diamantino, F., Voutsen, O., Cunha, M. S., Barroso, R., Lopes, M. J., & Carreiro, H. (2011). Incontinentia pigmenti in the neonatal period. BMJ case reports, bcr0120113708.



