Variant normalization is essentially reducing the representation of a variant to its canonical representation. Variant normalization ensures that the way a variant is represented is parsimonious and left-aligned and can also refer to splitting variants into their allelic primitives. VarSeq normalizes variants by default, but we offer users the option to forego one or more aspects of variant normalization. This blog will highlight situations in which a user might need to choose whether to split multinucleotide variants into their allelic primitives on import of a VCF into VarSeq.
Multinucletide polymorphism (MNP) describes the clustering of two or more adjacent variants on the same haplotype in an individual when represented as one complex variant. In VarSeq, the option to “Split features into allelic primitives” is selected by default. However, we are aware that this type of variant normalization can cause discrepancies in variant representation for some users, and some users may want to leave this box unchecked (Figure 1).
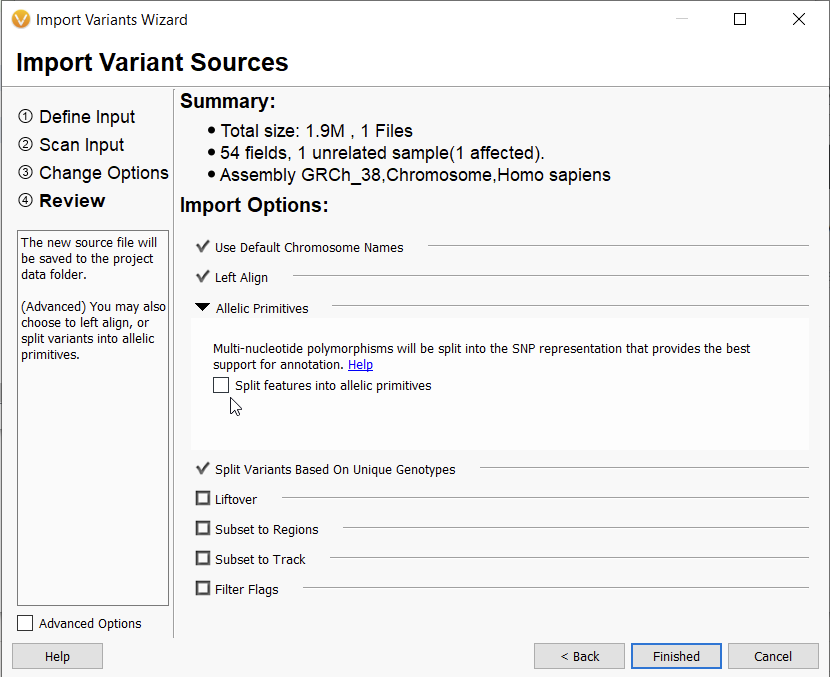
When the variants in an MNP are sufficiently close, such as being found within the same codon, the functional impact of that complex variant may be different from the impact of each variant when considered individually. We have observed, and others report, for example, in this article, that the majority of databases used for variant annotation are more likely to represent MNPs in their allelic primitives. Hence, VarSeq does this by default. However, a user may be concerned about missing a potentially relevant variant or, on the other hand, calling false positives, and will have to choose whether or not to split MNPs into their allelic primitives. An example of a discrepancy in variant classification is seen below, where an MNP is given a classification of likely benign while its individual variants are considered benign, the difference being the MNP fulfills the PM2 criteria on the basis of it being missing from the population frequency catalogs.

Variant normalization may affect the outcome of a user’s variant analysis is when filtering on sample-specific parameters such as read depth or zygosity or on population frequency catalog filters such as alternate allele frequency. In some instances, an applied threshold may retain an MNP but filter out one or more of the variants when a user opts to split into allelic primitives or vice versa. Luckily, VarSeq’s built-in Genome Browser allows you to visually capture both types of variant representation on manual inspection (Figure 3). This may be useful, for example, when investigating variants in a gene known to be associated with disease or when seeking to understand why a particular variant might not have been observed after filtering.
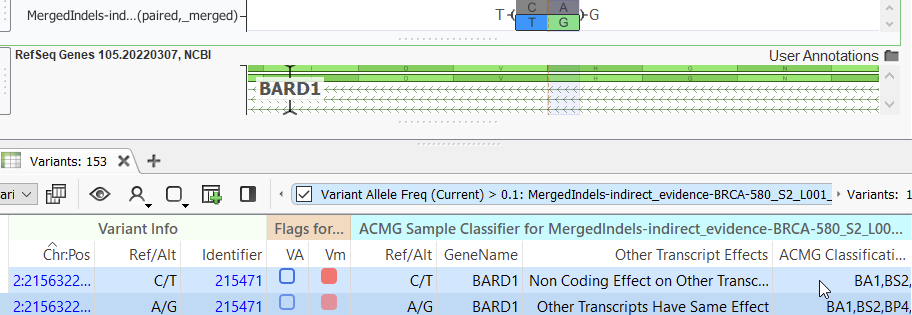
A user must be aware that the option to split or not split into allelic primitives will be applied across the board to all the variants imported into that particular project. Moreover, VSClinical, by default, will split variants into their allelic primitives. This often explains why the final numbers of filtered variants in a variant table sometimes differs from the final number of variants brought into a VSClincal evaluation.
Choosing whether or not to split MNPs into their allelic primitives is a complex topic and decision. With VarSeq, a user has the flexibility to compare and ultimately decide which representation they would like to use on a per-project basis. For additional information on variant normalization in VarSeq, please check out some of our other blogs on this topic. For any questions regarding this topic, please reach out to [email protected].
Golden Helix has developed innovative tools for the clinical interpretation of variants. VarSeq is developed with customizability and flexibility in mind. This way, it can work on a per-project basis and empower users. Please click the VarSeq icon below to learn more.




{kind=link}