To say the announcement of Dan MacArthur’s group’s release of the Exome Aggregation Consortium (ExAC) data was highly anticipated at ASHG 2014 would be an understatement.
Basically, there were two types of talks at ASHG. Those that proceeded the official ExAC release talk and referred to it, and those that followed the talk and referred to it.
Why is this a big deal? Well, since they have generously published their data not only in a great online browser but also in a downloadable VCF file, let’s take a look at the data and see for ourselves.
Population Catalogs
ExAC is the latest addition to a class of key public resources for doing variant analysis: the population catalog.
Population catalogs can have different goals, but they all provide the ability for a set of variants from your own samples to be annotated with the critical information of which variants are rare, common or are of different ethnicity and origins.
We spend a lot of time curating population catalogs, because knowledge of a variant’s prevalence is a very powerful filtering criteria. Getting their raw data from these catalogs converted into well formatted fields makes it easy to meaningfully bin your own variants, whether it’s on allele frequencies, germline versus somatic origins, or clinical significance.
Recent Updates
While ExAC has just arrived, the established variant catalogs have not been standing still. Here is a review of the existing catalogs and their recent updates. Note, all of these sources are available in the Golden Helix public annotation repository accessible from GenomeBrowse, VarSeq, and SVS.
NHLBI EVS/ESP (6500)
It didn’t seem that long ago that I remember the buzz at ASHG 2011 that the NHLBI where releasing 2,600 exomes through the HYPERLINK “http://evs.gs.washington.edu/EVS/” EVS server. Since then, multiple releases have brought the total number of exomes in this well annotated catalog to 6,500. These exomes are now included in ExAC! (Update: Dan MacArthur clarified that only a subset are included in ExAC due to dbGaP restrictions).
1000 Genomes Phase 3
While it has the comparatively small set of 2,500 individuals in its variant catalog, the 1kG project does sequence and call variants across whole genomes, bringing their total variant sites called in their latest Phase3 release to 79 million! The earlier Phase 1 variant set was called from 1094 individuals and contained 39.5 million unique variant sites.
COSMIC V71
COSMIC is a population catalog, but instead of cataloging germline mutations found in the population, it catalogs somatic variants discovered in cancer samples. With the integration of variants from The Cancer Genome Atlas, version 71 is the most compressive catalog of somatic mutations ever created and provides details about the tumor types and histology in which they occur. We also provide the gene level annotations COSMIC provides with relevant summary and curated oncology details.
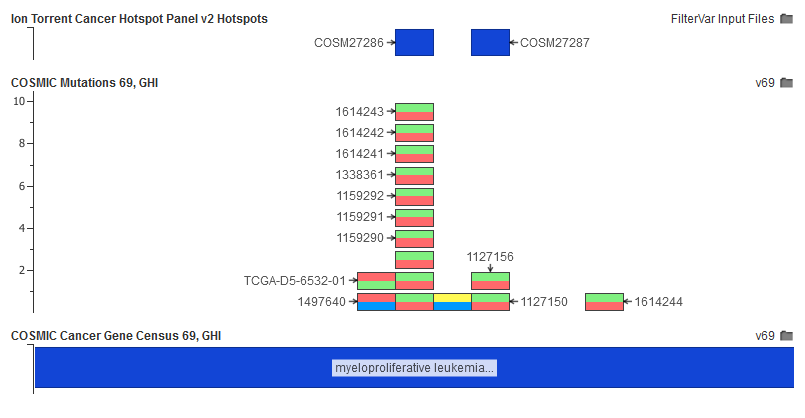
COSMIC breaks out each sample-variant pair into a record. We provide the fields in COSMIC with relevant hyperlinks and formatting to enable easy filtering.
dbSNP 142
Providing the critical services of enumerating unique variants with accession numbers in the form of RS IDs, the NCBI dbSNP database ends up integrating most other germline variant catalogs. The allele frequency information in the database is provided directly from 1000 genomes Phase 1 release, but dbSNP contains variants from many other sources as well (some of more dubious quality, but many flags are provided to weed these out).
ClinVar
With monthly data releases, NCBI ClinVar is an up-to-date repository of annotations about the clinical significance of variants as they relate to specific diseases. Many labs have contributed their archive of these classifications, and continue to submit on a regular basis their findings from ongoing clinical diagnoses. Nearly every gene with a known clinical trait association is well represented in ClinVar, and NCBI does an excellent job cross referencing each variant/disease classification with relevant clinical resources.
dbNSFP 2.7
While not strictly a population catalog, the database for Non-Synonymous Function Predictions does exhaustively enumerate every single letter substitution in the coding regions of the human genome that alters the protein sequence and annotates them with the output of many functional prediction and conservation score algorithms. The latest version has 9 functional prediction algorithms and 7 conservation scores applied to 90 million non-synonymous variants.
Not just for GRCh37
It’s impressive to see how quickly public annotations have become available for GRCh38, released just this last December. Care must be taken when creating population catalogs for GRCh38, because variant sets generated from NGS data are aligned and called against a specific reference assembly.
While 1000 Genomes has no plans (last time I had a chance to ask them) to re-align and call variants in GRCh38 coordinates, dbSNP has remapped all their cataloged variants (with the 1000 genome frequencies attached) since build 141. Similarly, the NHLBI variant set and dbNSFP has been “lifted over” to GRCh38 in their latest release.
An Order of Magnitude Bump
But what is crazy about the ExAC variant set is the absolute magnitude of their sample set. A whole order of magnitude larger than the previously largest exome catalog, it promises to expand our list of known variants dramatically… and it absolutely does.
The first thing this size of a cohort affords is a diversity of distinct population sub-groups. In this case, the bioinformatics team used PCA to stratify samples into the 7 population groups of:
- AFR: African & African American
- AMR: American
- EAS: East Asian
- FIN: Finnish
- NFE: Non-Finnish European
- SAS: South Asian
- OTH: Other
For each of these groups, there is a set of fields for each variant that provides the following informative counts:
- Alt Allele Counts: Counts for each observed alt allele
- Chromosome Counts: Total chromosomes called (usually 2xSampleCount)
- Homozygous Alt Counts: Number of samples with homozygous genotype
- Heterozygous Alt Counts: Number of samples with a heterozygous genotype
What this means is that you can, for example, ask if there are any variants in NGLY1 that are homozygous in Americans.
VEP Included
The last field of the 2.9GB compressed ExAC VCF is the very large “CSQ” field containing consequence predictions from Ensemble Variant Effect Predictor (VEP) for each variant. This field is a comma-delimited list of predictions per-alternate-transcript pair, with each field of the predictions delimited by ‘|’ and list items within those delimited by ‘&’. Got that?
With such great content in there, such as per-transcript SIFT/Polyphen predictions and the VEP provided sequence ontology and HGVS nomenclature, we thought it would be worth doing the work to make this dense, triple delimited field used as an annotation and filtering source.
So that’s what we did. We are going to release alongside the ExAC Variant Frequencies source containing all the frequencies and counts I described above, a second ExAC VEP Annotations source that breaks out all 53 million alternate-allele/transcript pairs into well formatted and “listified” fields with hyperlinks and filterable categories. This essentially allows you to have VEP annotations very cheaply for all known variants.
Yes, we provide all these annotations through our own algorithms and from other population catalogs directly, but as always in research, it never hurts to have multiple independent data points.
VarSeq Chomps on ExAC for Lunch
As I promised at the beginning, let’s actually do some analysis on ExAC to see what the most expansive variant population catalog in the world looks like.
If you haven’t heard before now, Golden Helix just launched VarSeq 1.0, and as a variant annotation and filtering tool designed to scale for handling thousands of exomes. Naturally, I fired it up and imported the ExAC variant set. Note that I did this whole analysis, from import to annotation, filtering and some investigation on my desktop computer over the course of my lunch yesterday.
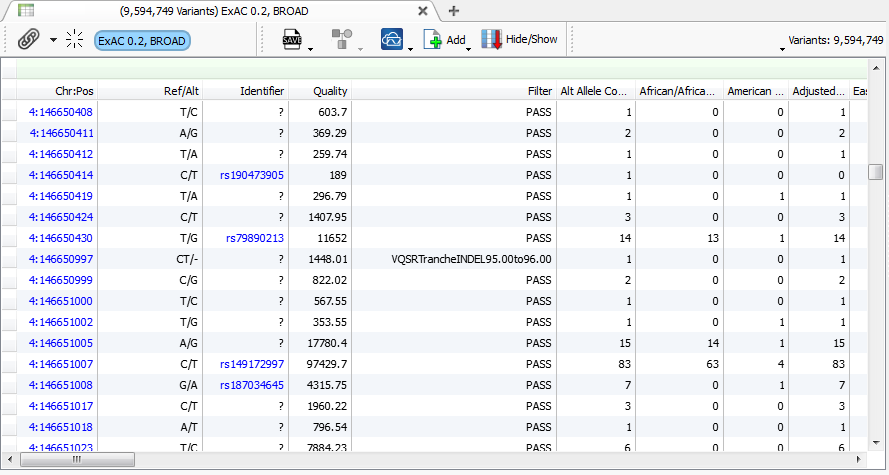
9.6 million variants from ExAC imported into VarSeq
Keep in mind the uncompressed VCF that represents all the data from this table I’m able to scroll through effortlessly on my desktop is 20GB in size!
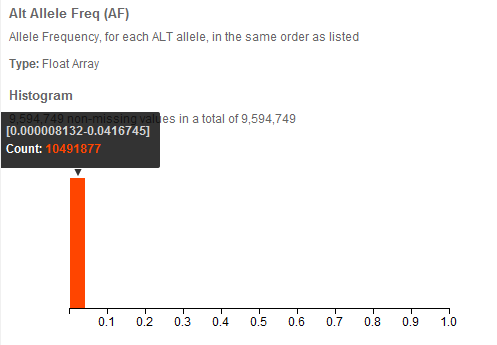
From the histogram of the Alt Allele Frequency column created by simply clicking on it, we are not surprised to find that very rare variants dominate the dataset.
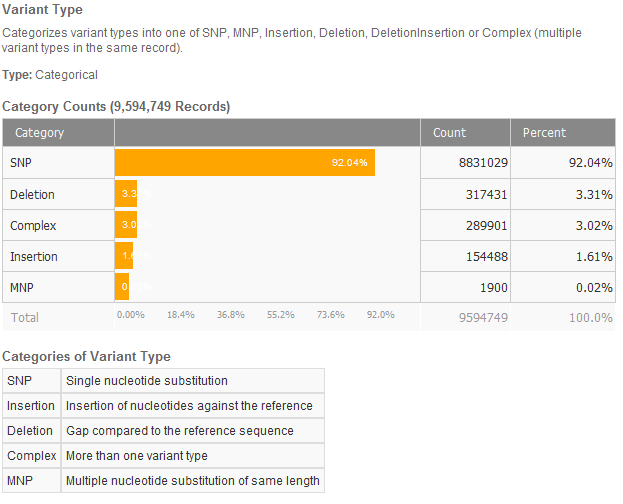
Using VarSeq’s algorithm catalog, I ran the Variant Type algorithm and in less than 30 seconds, had a breakdown of the types of variants in ExAC.
And finally, to really get a sense of how many of these millions of variants we have seen in the population catalogs I mentioned above, I annotated against each one and then added filters to the VarSeq filter chain to get a list of all variants seen in any of these. Note that each annotation took 2-3 minutes and this all occurred on my modest desktop without exceeding 1.5GB of RAM. This is laptop-duty work for VarSeq.
I added two thresholds to the frequency based population catalogs to point out that nearly all the annotated variants are below the 5% frequency threshold. dbSNP contains the most known variants.
But what is amazing about this analysis is that 6,637,439 variants in ExAC have not been cataloged in any population catalog.
In essence, ExAC has just tripled the number of known variants in coding regions of the human genome!
Finally, I thought it would be interesting to investigate those 5,670 variants in ExAC that have a Pathogenic clinical significance annotated in ClinVar. After sorting on allele frequency, it might be surprising to find that some variants marked as “Pathogenic” in Clinvar have frequencies close to 1. This finding highlights the importance of not assuming that the reference allele is always the clinically neutral allele!

VarSeq being used to investigate the 5,670 pathogenic variants in ExAC sorted by their Alt Allele Frequency
Invaluable in Unknowable Ways
I can’t imagine what this catalog means to all the researchers out there studying human genetics.
I know that while I was sitting in the ASHG session when ExAC went live, I quickly forwarded the gene-detailed page on NGLY1 on to Matt Might, who after having his son diagnosed with NGLY1 deficiency has initiated an amazing global network of family advocates for the disorder and has drawn the attention of the national press and was even plugged in the ASHG presidential address. Constantly in search of a better understanding of this diseases prevalence, Matt was amazed to see the details of the loss-of-function variant he carries and passed on to his son.
And that’s my two SNPs.
Most impressive Gabe!
I can’t wait to learn how to use this technology.
Dave
Great addition to SVS/VarSeq! This was tricky work.
Dear Gabe: Your case method of analysis is a great teaching aid in those of us, technically challenged in the SVS/VarSeq field. I enjoy reading all your articles: concise, direct with actionable steps from your conclusions.
That being said, is it possible to expand in Golden Helix’s algorithms the number of inputs i.e. nucleotide variables such as inosine (I) (IMP) and xanthine (X) (XMP) and Orotate (OMP). I am quite certain much of the ambiquity and variation from different data sets and populations will be clarified.
My 15 years of research as focused on proving these nucleotides are not only important but necessary for faithful DNA replication to occur. If you look at my colored coded work you will notice the inosine family (orange) is critical to purine metabolism. i.e. Adenine (red) and Guanine (green) the current purine codons and anti-codons in the DNA and RNA genetic codes cannot replicate DNA without IMP (the first closed purine nucleotide) , HPRT (purine salvage), and xanthine oxidase (purine catabolic degradation.
No software on the market allows for expansion of the ATGC or AUGC alphabets. It would be very interesting to allow expanded nucleotide inputs to be correlated with GA and AG mutations. The A to I mRNA editing and alternative splicing is part of the revolutionary small mRNA i.e. 22-29 mer research spreading across the field.
Keep up the good work and I work be happy to work with you on this new product development project for nucleotide expansion. Thanks for your eloquent writing and hopefully your visionary openness to my research. Sincerely John A Berger Novagon DNa
Pingback: In Pursuit of Longevity: Analyzing the Supercentenarian Whole Genomes with VarSeq | Our 2 SNPs…®
Pingback: Accurate Annotations: Updates to the NHLBI Exome Sequencing Project Variant Catalog | Our 2 SNPs…®
Pingback: VarSeq is a better ANNOVAR, snpEff and VEP | Our 2 SNPs…®
Pingback: Genes to Genomes: a blog from the Genetics Society of America
H Gabe,
In ExAC browser For the variant given below the Homozygous count is shown in the table
1 55512194 . A C
But for another variant given below Heterozygous count is shown
Y 27001073 . T C 180.10 AC_Adj0_Filter
Kindly see a sample data taken from ExAC VCF file .How can we identify that below data is Homozygous or Heterozygous?Depending upon what criteria Homozygous/ Heterozygous is displayed in the ExAC browser table
1 55512194 . A C 2038.69 PASS AC=1;AC_AFR=0;AC_AMR=0;AC_Adj=1;AC_EAS=0;AC_FIN=0;AC_Het=1;AC_Hom=0;AC_NFE=1;AC_OTH=0;AC_SAS=0;AF=8.236e-06;AN=121412;AN_AFR=10398;AN_AMR=11574;AN_Adj=121372;AN_EAS=8650;AN_FIN=6614;AN_NFE=66716;AN_OTH=908;AN_SAS=16512;BaseQRankSum=-1.041e+00;ClippingRankSum=0.152;DP=2157434;FS=3.206;GQ_MEAN=78.74;GQ_STDDEV=19.80;Het_AFR=0;Het_AMR=0;Het_EAS=0;Het_FIN=0;Het_NFE=1;Het_OTH=0;Het_SAS=0;Hom_AFR=0;Hom_AMR=0;Hom_EAS=0;Hom_FIN=0;Hom_NFE=0;Hom_OTH=0;Hom_SAS=0;InbreedingCoeff=-0.0000;MQ=59.54;MQ0=0;MQRankSum=-4.850e-01;NCC=0;QD=9.99;ReadPosRankSum=1.11;VQSLOD=0.573;culprit=QD;DP_HIST=0|19|34|20|4290|15500|14027|9890|7216|4238|2432|1301|708|360|191|119|83|64|56|158,0|0|0|0|0|0|0|0|0|0|0|0|0|0|0|0|1|0|0|0;GQ_HIST=0|0|0|1|32|8|15|13|6|5|5|7|17461|8101|3904|5743|4890|2544|3855|14116,0|0|0|0|0|0|0|0|0|0|0|0|0|0|0|0|0|0|0|1;CSQ=C|ENSG00000169174|ENST00000543384|Transcript|intron_variant|||||||1||1|PCSK9|HGNC|20001|protein_coding|||ENSP00000441859||F5GWF0_HUMAN|UPI000206501F||||1/9||ENST00000543384.1:c.-77-5757A>C|||||||||||||||||||,C|ENSG00000169174|ENST00000452118|Transcript|splice_acceptor_variant|||||||1||1|PCSK9|HGNC|20001|protein_coding|||ENSP00000401598||B4DEZ9_HUMAN|UPI00017A6F55||||2/5||ENST00000452118.2:c.400-2A>C||||||||||||||||INTRON_SIZE:2488|||HC,C|ENSG00000169174|ENST00000302118|Transcript|splice_acceptor_variant|||||||1||1|PCSK9|HGNC|20001|protein_coding|YES|CCDS603.1|ENSP00000303208|PCSK9_HUMAN||UPI00001615E1||||2/11||ENST00000302118.5:c.400-2A>C||||||||||||||||INTRON_SIZE:2488|||HC
Thanks,
Amruta Nambiar
Hey Amruta,
The first variant you mentioned only occurs as a single het with allele count of 1. Here it is in ExAC:
http://exac.broadinstitute.org/variant/1-55512194-A-C
And we display the same information in our track.
There is per-field documentation in the VCF header that describe the INFO codes. For example in this variant Het_NFE=1, which means the single het genotype is in the “Non-Finnish European” cohort.
The second variant has an adjusted allele count of 0 (AC_Adj=0), meaning that with filters applied they are not confident it is present in any samples:
http://exac.broadinstitute.org/variant/Y-27001073-T-C
One thing we provide that is not in the VCF file is a computed “Adjusted Alt Allele Freq”. This is what is displayed in exact, but they only have the un-adjusted “AF” field in the VCF.
The adjusted alt allele frequency is just the AC_Adj / AN, which in this case is 0 / 200 = 0.0
Hi Gabe,
How can an alternate allele have a frequency of 0.994 ? if so, it must have been annotated as the ref allele right?
Thanks in advance,
Ajitha
The reference allele is set by the genome reference. When an alternate has a frequency greater than 0.5, it is actually the “major allele”. Some of these “ref is minor” alleles have been flipped in GRCh38, but are always possible in different populations.
Gabe
Hello Gabe, thanks so much for the article!
I’ve got a question about ExAC data, probably I don’t get something.
So in the web-site they said that 1000 genomes cohorts are included, but some of the SNPs , for example rs17047586 is presented in 1000 genomes, but I can not find it in Exac.
Do you know why could it happen?
ExAC is a based on exome sequencing and so will be missing many variants that are intronic or intergenic.
The RSID you mentioned is 100kb into the intron of FAM19A1 and so will definitely not overlap the exome capture kits regions of coverage.