DNA methylation is becoming more relevant as a clinically important biomarker, and long-read pipelines are making it easy to get this information in the same sequencing run as small variants and larger structural variants. Even though there are no official guidelines for addressing DNA methylation, it is still useful to analyze and evaluate this data, so we would like to highlight the ways in which VarSeq can be used to analyze DNA methylation.
Tests to determine methylation status of genes have been used for many years in the diagnosis of imprinting disorders and to identify cancer biomarkers based on methylated DNA. Array-based, NGS-based, and now long-read third-generation sequencing technologies have increased the range of methylation testing and improved the utility of these genetic tests for diagnostic and prognostic classification of tumors — complementing somatic analysis workflows where methylation biomarkers such as MGMT are clinically actionable. Though the research supports the utility of this data type, there are no current standardized guidelines for implementing this technology or interpreting the DNA methylation data in a clinical setting. However, the ACMG and other working groups are discussing the implementation of DNA methylation arrays for clinical diagnostics.
In the spirit of getting ahead of the game, here are a few ways to leverage VarSeq to analyze your methylation data.
Import Your DMR Files
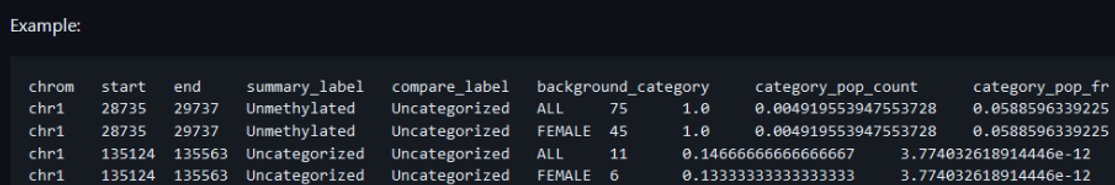
PacBio and Oxford Nanopore sequencing pipelines produce global profiles of DNA methylation alongside the standard sequencing output (Figure 1). Differentially methylated regions (DMR) can be derived from this data and imported into VarSeq for filtering and visualization in Genome Browser. Conceivably, differentially methylated region files of a similar format from any pipeline can also be imported.
Report Methylation Biomarkers
As mentioned, there are no guidelines for interpreting DNA methylation cancer biomarkers. However, in VSClinical, we do enable users to create and, if desired, report user-defined interpretations regarding methylated DNA biomarkers as part of a broader clinical interpretation workflow — including biomarkers such as MGMT methylation for glioblastoma.