In the last installment of this series, I covered the basics of variant interpretation and how it fits into the genomic testing process. Now we can cover in more detail how VSClinical works, what algorithms and annotation sources power the recommendations and how the ACMG criteria are organized into useful categories.
VSClinical is built to make the process of evaluating variants based on the 33 criteria of the ACMG guidelines efficient, less prone to differences between evaluators and more enjoyable. This requires leveraging the full arsenal of algorithms and capabilities of VarSeq, and quite a few new ones we have built specifically to support this product.
What Powers VSClinical
VarSeq has matured as a powerful and flexible annotation and filtering platform for NGS small variants and CNVs over the years by working closely with researchers and clinicians to support their many use cases. To power the VSClinical auto-computed scoring recommendations and context-specific evidence, many VarSeq algorithms and carefully curated annotation sources are leveraged. Here is a review of some of them and how they are presented and visualized.
Allelic Matching Population Catalog Annotation:
VSClinical automatically annotates gnomAD and the 1000 Genomes with genotype counts. Both of these sources have specialized curation steps to prepare their raw format to be optimized for this use case. VSClinical automatically displays the sub-population with the highest frequency for consideration.
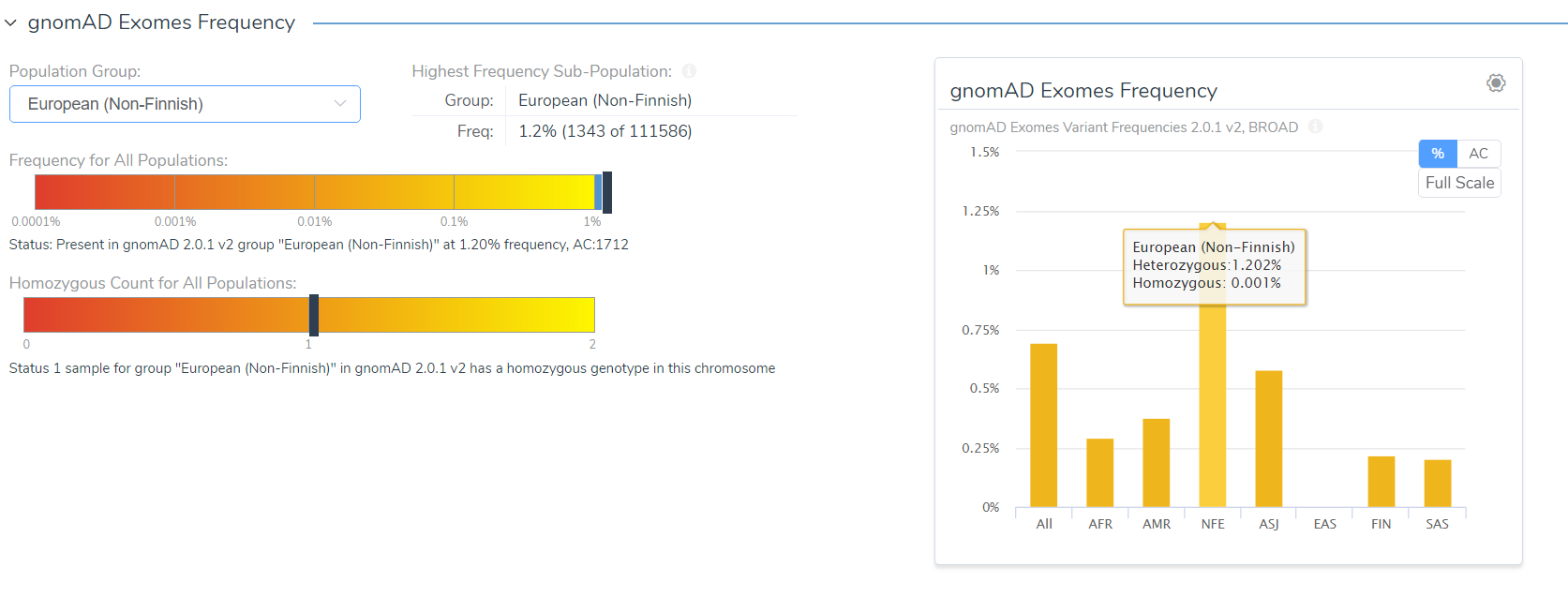
Along with a log-scale of the lower parts of the frequency spectrum, VSClinical has a dedicated visualization for the number individuals have homozygous genotypes for a given variant. When interacting with the frequency data, the graph on the right can be switched from displaying percentage to total allele count (AC) per population. Homozygous genotypes are colored in red in this graph, with specific counts available on hover. These details help answer scoring questions about whether a variant occurs at a higher-than-expected frequency for a disorder and whether it ever occurs in a homozygous state in healthy individuals.
OMIM Phenotypes and Genes
To support a frequency filter threshold that matches the inheritance model and severity of known disorders, VSClinical presents the specially curated OMIM-linked phenotypes for each gene with a built-in viewing of the details.
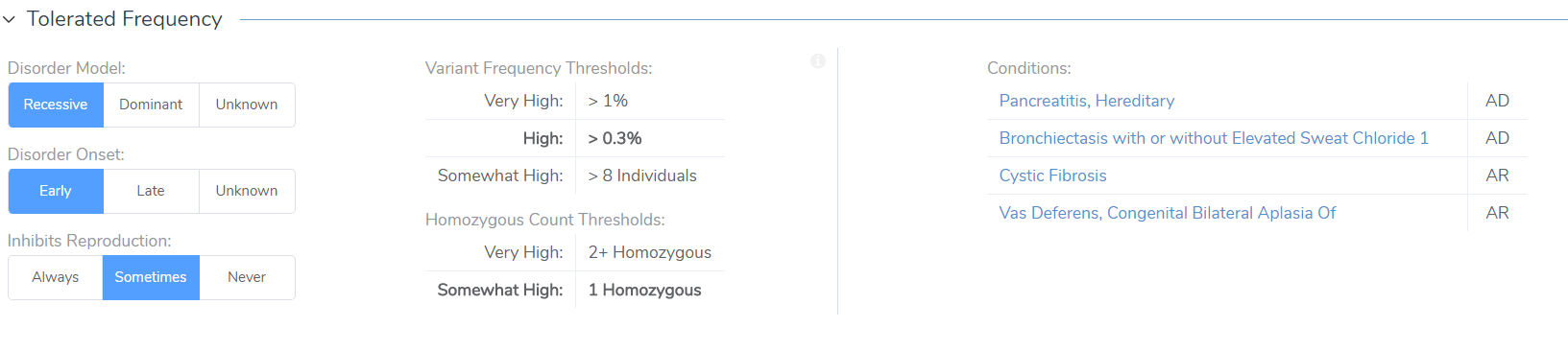
The settings on the left will adjust the threshold and count used for the relevant scoring criteria’s recommendation. The OMIM phenotypes are also used to auto-complete the input of the disorder or condition name when interpreting the variant.
Genes with Default Transcript Section
The interpretation of a variant must be in the context of a transcript, and in our upcoming VarSeq 2.0 release, we will be updating the selection of the “Clinically Relevant” transcript to closely match the transcript reported by other clinical labs that have submitted to ClinVar. While all analysis in VSClinical is computed on the current transcript, you can switch the interpretation over to any other overlapping transcript on the fly. A warning will be displayed when there are other transcripts that have a different gene-level effect, giving you a chance to evaluate their significance.
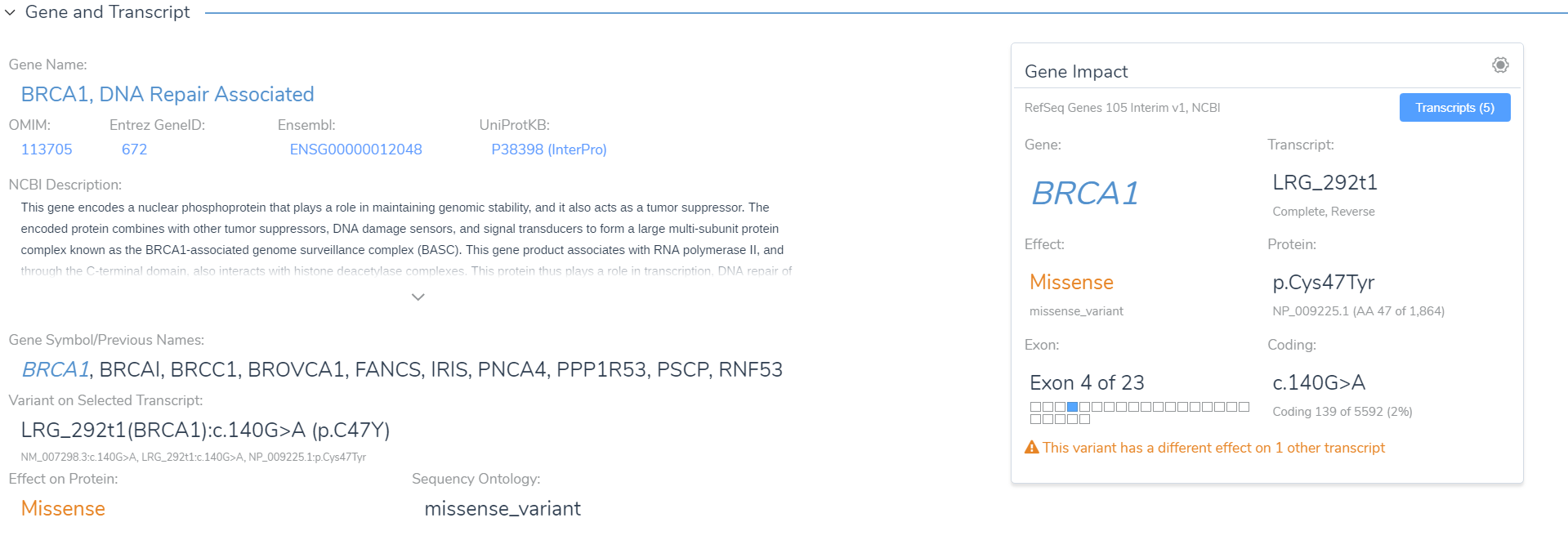
Along with our variant-transcript details, other useful gene resources are presented for the current gene including previous aliases and the very popular NCBI gene summary.
Existing Variant Interpretations for a Gene
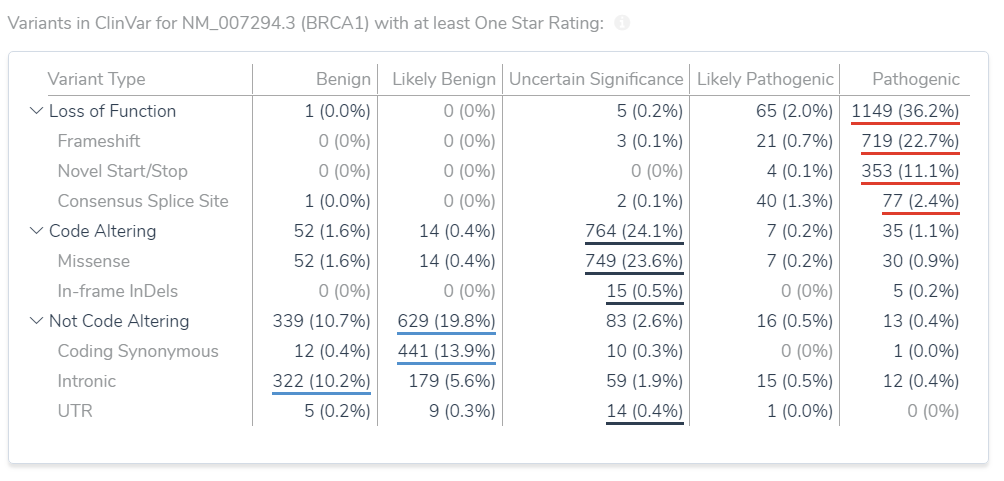
A few critical questions for the ACMG guidelines require understanding what types of variants have been established to Pathogenic or Benign in the current gene. For example, are loss-of-function variants like frameshifts a known mechanism for disease? This is usually the case, but there are exceptions, like MYH7. For a given gene, this can be determined by looking at all the frameshift variants in ClinVar to see what classification has been predominantly assigned. For other questions, it is useful to do a similar analysis for missense variants. VSClinical summarizes this information into a helpful table grouped by variant type.
Given this table about BRCA1, its clear one has to be careful when evaluating a missense mutation as just as many have been classified Benign as Pathogenic.
While a gene summary provides some useful background on the possible mechanism of disease for a gene, more weight should be placed on what has been established for variants in the immediate vicinity as a variant of interest.
Here we present a table centered around a variant with the first column showing the relative distance of other variants to the variant being evaluated. The table can be filtered the type of variant, the established classification, the location and the ClinVar star rating (review status).
Now you can easily answer questions like “Are there any two-star rated missense variants classified Pathogenic within 10 amino acids of my variant?” or “How many benign Loss of Function variants are in the same exon as my variant?”. Most importantly, the criteria PM1 requires evaluating whether a variant is in a mutation hot-spot, which can be answered by looking at the 5 amino acid window (or highly conserved functional domain) around the current variant.
Tolerate of Loss of Function or Missense Scores
Along with looking at rate and prevalence of specific types of variants submitted to ClinVar, the rate and frequency of variants in large population cohorts can also be used to understand what types of variants are tolerated by a gene.
The ExAC project computed a model for determining the expected frequency rate of missense and loss-of-function mutations for each gene. That expected rate is compared to the observed number of variants of each type per gene from the 64,000 individuals sequenced. The result is per-gene score based on the statistical likelihood of seeing the difference between the observed and expected by chance. For loss-of-function variants, we present this “Intolerance of Loss-of-Function” factor (pLI) factor as supporting evidence of the PVS1 criteria. For addressing one of the key caveats of this criteria, we also present a visual depiction of the physical proximity of the variant to the final exon and the 50nt coding sequence preceding it. Inside these regions, frameshift variants may not truncate enough of the protein to result in complete loss of the protein product through nonsense-mediated decay.
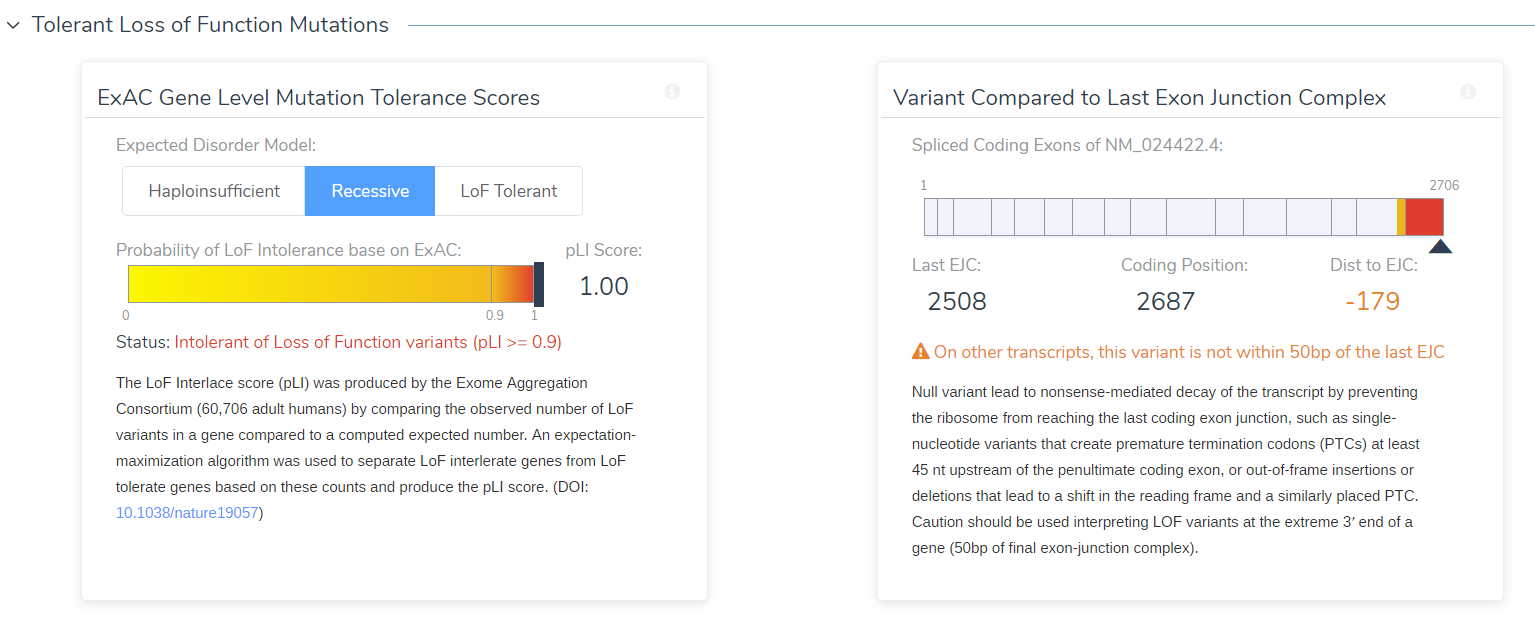
A similar score is computed and presented for missense variants. Based on recent popularity and requests, we also specially curated the Missense Badness and MCP scores, which capture when a given region is depleted of missense mutations with evidence for increased deleteriousness. Note though that the second score MCP is a combined, or ensemble score of Missense Badness with Polyphen-2, and as such should be considered an orthogonal piece of computational evidence.
Coding Sequence
One of the most helpful ways to visualize a variant is to see how it changes both the coding sequence and the resulting amino acid residues in the mutated protein product. This is critical in answering questions about in-frame insertions and deletions, such as whether they are part of an amino acid repeat region or involve more unique residues.
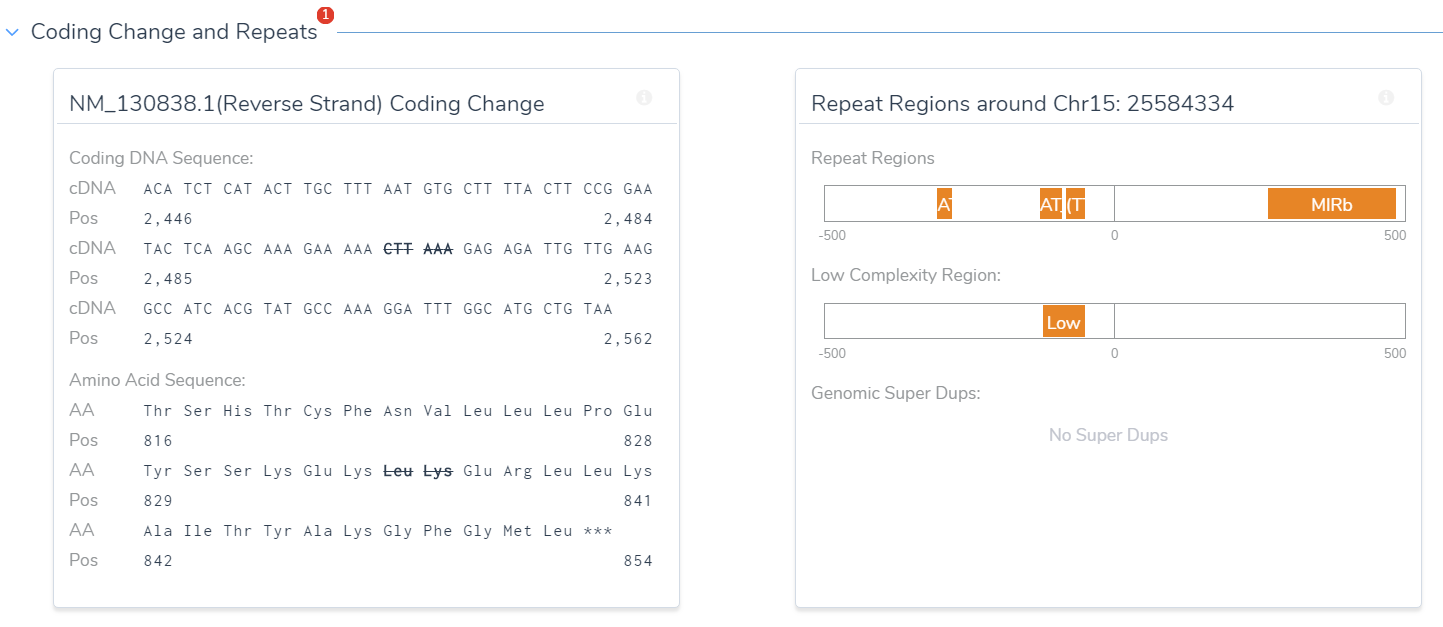
Here a the deletion of 6bp results in the in-frame removal of two amino acids near the end of the protein sequence. Although there are repeat regions nearby, the deletion does not occur in a repeat.
Although specifically designed for length polymorphism, it can be helpful in judging the impact of frameshifts as well:
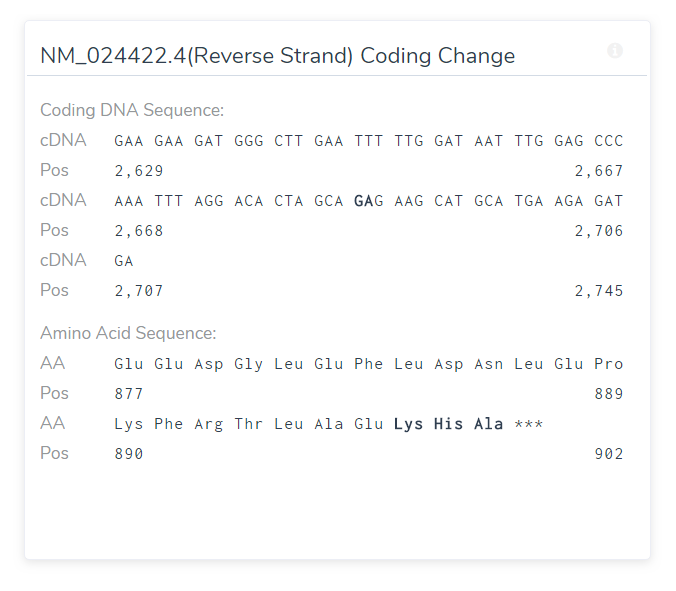
A frame-shift variant that inserts a GA results in the change of three amino acids followed by a stop codon.
Functional Prediction and Splicing
Our Computation Evidence section provides a number of exclusive-to-VSClinical algorithms including our implementation of SIFT and PolyPhen2 that run directly on the Multiple-Sequence-Alignment and our custom implementation of four splice site algorithms. These algorithms require their own dedicated discussion so we will pick them up in our subsequent posts.