
In the upcoming release of VarSeq 2.6.2, we have added the ability to force call reference alleles using the BAM files associated with the sample. This feature extends the current force call functionality, which allows filling in reference alleles from GVCFs. This is an important option to enable when running pharmacogenomics pipelines with VarSeq, as it allows for inferring the reference status of alleles used by the PGX haplotype caller. As you may recall, each haplotype considered by VarSeq is composed of a set of alleles. When the sample has all of the alleles in a haplotype, the haplotype can be assigned to the sample. The set of haplotypes assigned to a sample is then used to determine individual drug sensitivity, among other attributes. Unfortunately, when variant calls are missing at sites required for the haplotype, haplotype assignment can’t be completed. To overcome this issue, VarSeq has a couple of options for filling in calls at these locations.
These options are:
- Force calling the alleles from GVCF regions
- Force calling the alleles as reference
- Force calling the alleles using the sample coverage
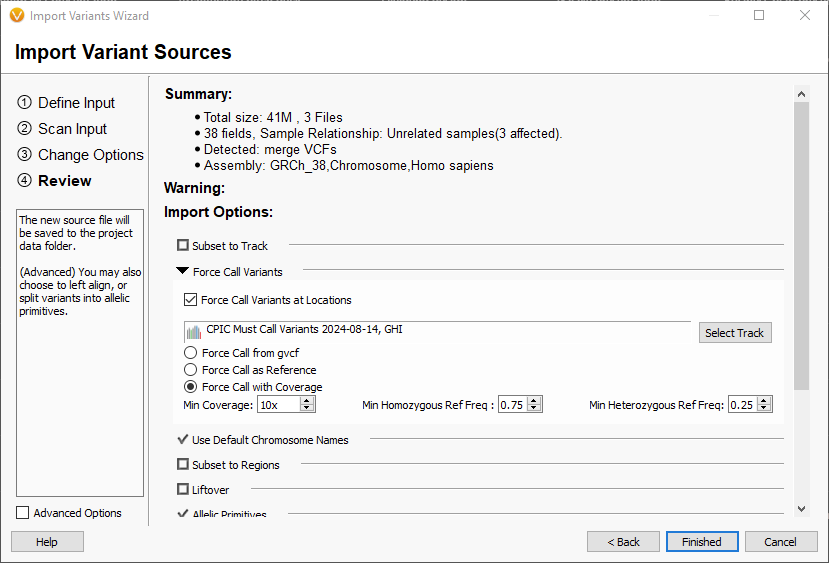
Each of these options is driven by the force call track selected which defines the union of all of the alleles in all of the haplotypes considered by VarSeq. Today we are focusing on the “Force Call with Coverage” option. When you select “Force Call with Coverage” VarSeq will try to fill these required variants by using this input priority:
- Check if the variant was called and is in the input VCF file.
- Check if the variant overlaps a GVCF span from the input VCF; if so, use this to assign a reference genotype.
- Read the sample coverage for the variant region in the associated sample BAM file, if there is adequate coverage and percentage of reference reads to non-reference reads meets the thresholds call the variant as reference.
This will allow calling of reference alleles when you are unable to adjust your secondary variant calling pipeline to incorporate the “must call variants” track, and your pipeline doesn’t output variant calls with GVCF spanning records. Here is an example of a reference call filled from the BAM file:
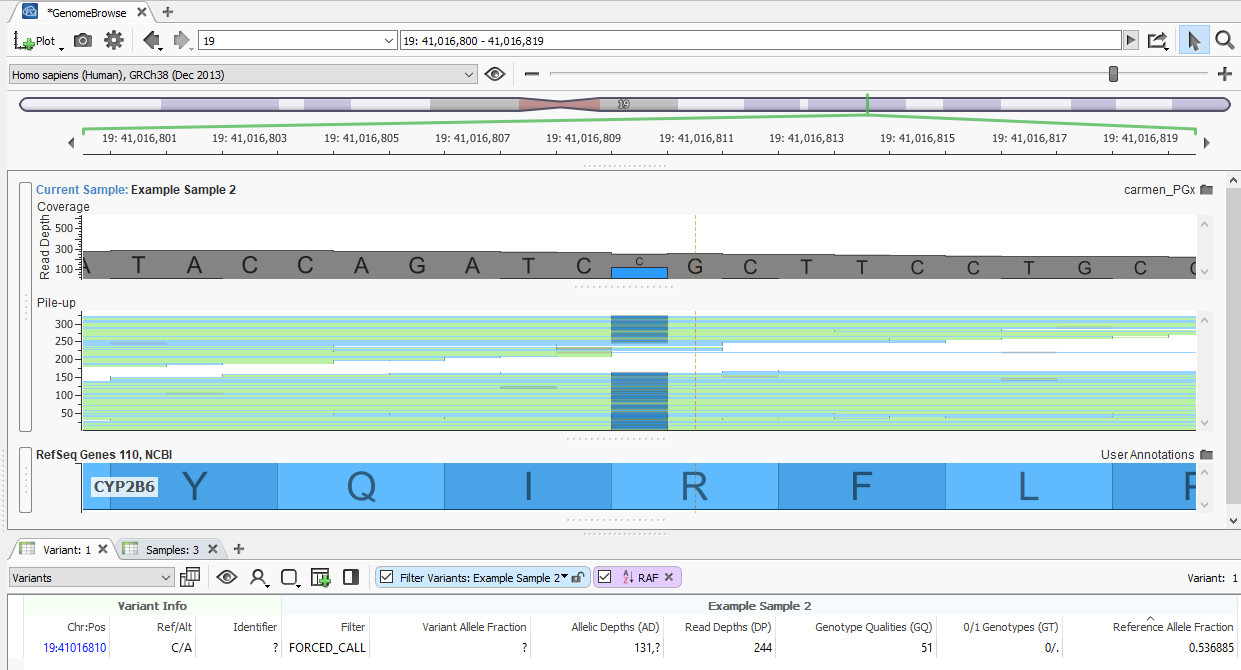
There are a couple interesting things to note when looking at this feature:
- The Filter field has the FORCED_CALL value. This indicates that this variant was added because there was a record for it in the force call track selected on import. This flag can be helpful when trying to determine which variants were added by forcing calls, and which variants were in the original VCF.
- The Reference Allele Fraction is 0.536; this is calculated by dividing the number of reads for the reference allele (Allelic Depths – 131 ) by the total depth (Read Depths – 244). This new field is computed for all of the variants force called from the BAM file; the AD and DP fields are also filled in for these variants by reading from the BAM file.
- The 0/1 Genotypes field has a half call (0/.) for the reference allele. This is a half call because the Reference Allele Fraction is above the heterozygous call threshold of 0.25 but below the homozygous call threshold of 0.75. These thresholds can be adjusted in the settings on the final page of the import wizard.
Overall, this new feature will allow the PGx Variant Detection and Recommendations algorithm to confidently call sample haplotypes independent of the secondary pipeline choices. You can look forward to this feature and many more in the upcoming release of VarSeq 2.6.2.
Please check back soon for more updates on the upcoming release, and feel free to reach out to [email protected] if you have questions about this or any other new feature once 2.6.2 is available!



