A prerequisite for clinical NGS interpretation is ensuring that the data being analyzed is of high enough quality to support the test results being returned to the physician. The keystone of this quality control process is coverage analysis. Coverage analysis has two distinct parts.
- Ensure that there is sufficient coverage to be confident in called variants
- Make certain that no variants went undetected in tested regions due to an insufficient number of reads
In the past, computing coverage metrics was part of the secondary analysis (alignment and variant calling) portion of the clinical workflow. This is very sensible in terms of workflow efficiency and data management. Secondary analysis tools are already long-running pipelines which create a great deal of meta-data during the processing of sequence reads. Thus, it is sensible to include the creation of quality assurance metrics in this pipeline. In fact, both GATK and Ion Torrent provide tools to do this via their Depth Of Coverage Command and Coverage Analysis Plugin, respectively. In fact, I’d encourage all users to add these steps to their upstream, bioinformatics pipeline.
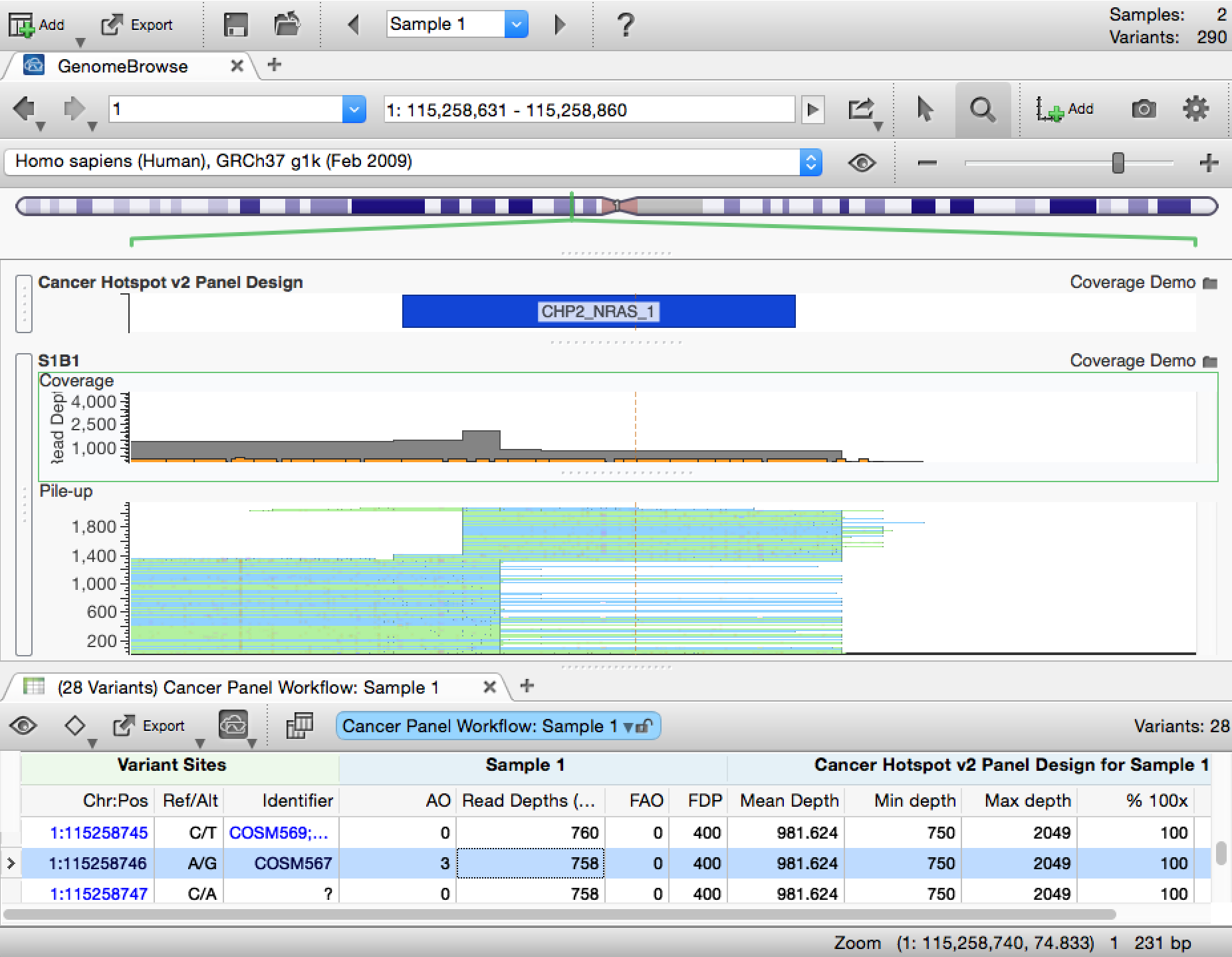
However, as genomics evolved and more and more groups have become involved in the analysis, it is increasingly difficult to track all the information generated by a single sample. We recognize that often coverage information is not included in the package of data that the clinician receives. Moreover, having coverage metrics available in the context of variant analysis simplifies the entire clinical interpretation workflow. To ensure that a complete picture is available for each variant, we are happy to announce that a Coverage Analysis algorithm has been added to VarSeq 1.2.1.
The only requirement to use the algorithm is that you have both the BAM file for the sample you are analyzing and a BED file defining the regions of interest. We then analyze these regions to determine the following statistics:
- Average depth of coverage
- Minimum read depth
- Maximum read depth
- Percent of bases in a region meeting different coverage thresholds
These computations are done in a strand aware manner, which can often help to uncover sequencing anomalies.
The metrics are available in two forms. First, each variant displays data about the region in which it resides. This binding allows variants from suspect regions to be flagged or filtered out, which can help to prevent false positives. Second, each region in the bed file can be examined. This mode of analysis ensures that all the targeted regions were sequenced, which is crucial to preventing false negatives.
To run the coverage stats algorithm, simply attach BAM files to each sample during import. Then select “Coverage Statistics” from the algorithm selector. You will then be prompted to provide a BED file. After clicking “Ok”, VarSeq will do the rest. Once the algorithm has finished running you’ll be able use the created fields to create filters or visualize coverage information using the automatically computed histograms in the data console. Like other algorithms, coverage statistics can be run from the command line using VSPipeline. Using this command line interface, it is easy to automate and precompute quality control steps, in addition to variant annotation and filtering.
We hope that bringing this small piece of the secondary analysis workflow into the interpretation phase will make it significantly easier to gain the confidence that is necessary in clinical testing environments. If you have any questions about how to use the algorithm or comments about how to make it even better, don’t hesitate to reach out to us at [email protected].