In trio workflows, one of the most important factors in scoring a variant is understanding how that variant is inherited from the parents. Likewise, when looking at extended families, the segregation, or presence of the variant among the affected versus unaffected individuals provides evidence for its pathogenicity for a given phenotype or disease. Given the nature of Copy Number Variants (CNVs) as large events being detected from variable depth Next-Gen Sequencing (NGS) data, it is difficult to consistently call the exact same start and stop positions between samples. For this reason, the inheritance and segregation of a CNV in a trio is not easily determined. To solve this problem, we developed a probabilistic algorithm that can provide confidence in whether a CNV was inherited, occurs de Novo, or segregates among affected individuals.
Importance of CNV Inheritance in the ACMG Guidelines
The inheritance and family history information of a patient can provide vital evidence for establishing the clinical significance of an observed Copy Number Variation (CNV). The recently published Technical Standards for the Interpretation and Reporting of Constitutional Copy Number Variants by ACMG and ClinGen recommend examining whether or not a given CNV segregates among similarly affected family members [1]. Such segregation can provide evidence supporting pathogenicity while discovering that the variant is missing from another affected individual or is found in an unaffected individual, can provide moderate to strong evidence against pathogenicity.
We are proud to announce that VarSeq now supports performing such analysis with our new Copy Number Probability and Segregation algorithm. This algorithm computes the expected copy number for each called CNV, while identifying other samples likely to share the variant and, for trio workflows, it computes the probability that the CNV is present in the mother or father.
Our copy number inference algorithm relies on a probabilistic model which is used to compute the most probable copy number for each target based on the target’s corresponding ratio and z-score values. For deletions, two samples are said to share the same event, if the inferred copy numbers for the targets spanning the event are the same. For duplications, two samples are said to share an event if there are more than two copies for the targets spanning the event.
The Copy Number Probability and Segregation Algorithm
Now that you know a little bit about how this algorithm works, let’s look at a hypothetical example designed to illustrate the utility of this method in the context of a trio analysis. Suppose we have a young boy presenting developmental delay and macrocephaly. His mother has a history of a partial thyroidectomy for a multi-nodulary goiter and was diagnosed with Cowden syndrome, as she also had hyperkeratotic pits on the palms of her hands, mild ptosis, and a large head circumference.
A trio exome analysis is performed which includes CNV calling using VS-CNV. After performing annotation and filtering, we identify two clinically interesting CNVs: one is a deletion of the last two exons of ATM, while the other is a heterozygous deletion of exons 3-5 of PTEN. The latter is shown in the plot below:
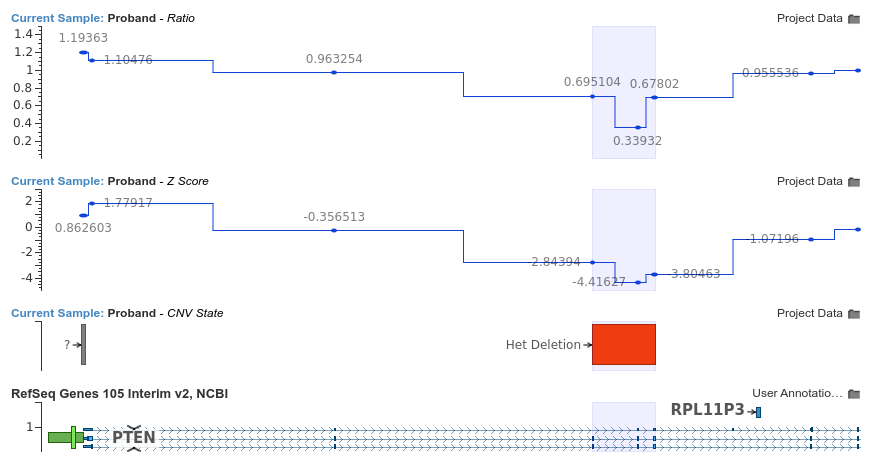
The z-scores and ratios for both of these events give us no reason to doubt the validity of either call. Next, we run VarSeq’s Copy Number Probability and Segregation algorithm to see if either mutation segregates with the mother or father. These results are shown below (click to enlarge):

We can clearly see from the segregation results that the mother shares a heterozygous deletion in this region of PTEN. Mutations in PTEN are known to be associated with Cowden syndrome [2] and the proband’s clinical features are consistent with this diagnosis [3]. Additionally, the clinical features of patients with PTEN mutations are often quite variable and presentations can differ significantly even within the same family [4]. While a single segregation, such as the one described here is not sufficient for a pathogenic classification, it does provide an important piece of supporting evidence for the variant’s clinical significance.
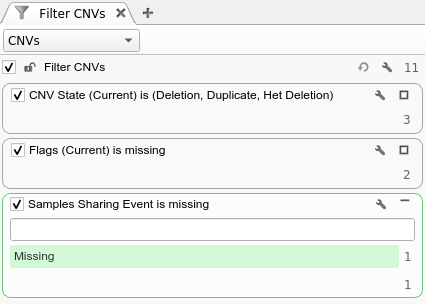
As you can imagine, another use case that can leverage the output of this algorithm is looking for de Novo CNVs. For every proband CNV, the probability of the same CNV will be evaluated in both parents, regardless of whether the CNV calling algorithm found an overlapping CNV event or not. With this output, it is possible to, for example, filter on the “Samples Sharing Event” column being Missing to find CNVs that are not shared. For more fine grained control, the user can filter on the Mother and Father Copy Number Probability columns with thresholds close to 0 to find CNVs with low probabilities of being in either parent. An example of the former de Novo filter chain is shown below:
Hopefully, this blog post has demonstrated the value of our new Copy Number Probability and Segregation algorithm. While there are numerous tools capable of calling CNVs from NGS data to my knowledge, none support the robust annotation, computational analysis, filtering, and clinical interpretation capabilities of VarSeq. If you have any questions or comments about the information presented here or about our software, please don’t hesitate to enter them into the comments below or email us personally at [email protected].
References:
- Riggs, Erin Rooney, et al. “Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen).” Genetics in Medicine 22.2 (2020): 245-257.
- Ueno, Yuichi, et al. “A novel missense PTEN mutation identified in a patient with macrocephaly and developmental delay.” Human genome variation 6.1 (2019): 1-4.
- Hanssen, A. M., and Jean-Pierre Fryns. “Cowden syndrome.” Journal of medical genetics 32.2 (1995): 117-119.
- Varga, Elizabeth A., et al. “The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay, and macrocephaly.” Genetics in Medicine 11.2 (2009): 111-117.



