Whole exome sequencing workflows using SNP & Variation Suite (SVS) was presented in a recent webcast, by Dr. Robert Hamilton from the Hospital for Sick Kids. In particular, he performed some filtering on his data to look for only heterozygous variants in his sample of interest, removed variants with allele frequency less than 0.005% based off of the ExAC Variant Frequency catalog and lastly performed some filtering based on coding classifications determined by the variant classification algorithm.
All of these variant annotation and filtering options can also be performed using our VarSeq software, and once the workflow is complete in VarSeq you can easily replicate the workflow on new samples using the available templating options. Below we will take a look at each filter options between the two products.
Filtering down to heterozygous variants can be done in SVS by going to DNA-Seq > Activate Variants by Sample Genotypes and selecting the zygosity for each sample of interest. This option works best for a small number of samples, if you have a large number of samples then we have an add-on script that can better handle this workflow that can be downloaded from the following link.
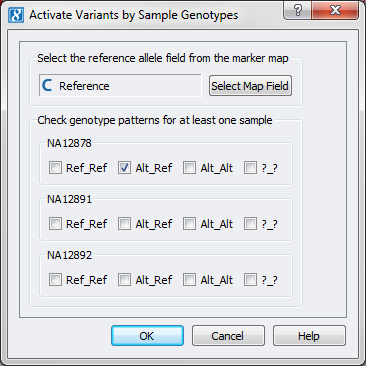
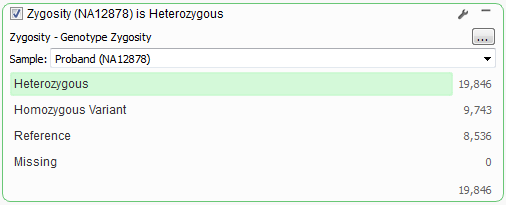
The same workflow within VarSeq requires first computing zygosity for your samples which can be done by going to Add > Computed Data and selecting Variant > Per Sample > Genotype Zygosity algorithm. Then create your filter card by right-clicking on the Zygosity column and selecting “Create Filter Card for this Column” and selecting your zygosity options. The card can be updated to a particular sample using the wrench icon options on the card. Once again this option is best if only filtering based on a small number of samples, if you have a large number of samples in your project then we recommend using the Add > Computed Data and then Variant > Project/Cohort > Count Alleles algorithm and then filtering on the provided project counts.
Next comes our rare variant filter based on the ExAC Variant Frequency catalog. Within SVS you will want to go to DNA-Seq > Annotate and Filter Variants and select the “ExAC Variant Frequencies 0.3, BROAD” annotation source then click Next to set your filter options.
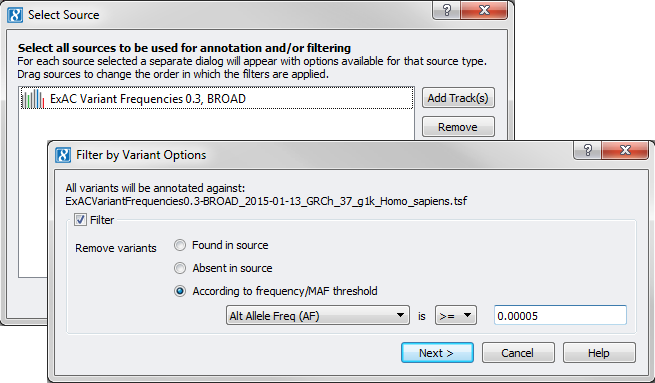
In VarSeq, you will want to first annotate by the ExAC source by going to Add > Annotation and selecting the source. Once the annotation is complete then right-click on the “Alt Allele Freq (AF)” column to create your filter card and set your thresholds accordingly.

The last part of the workflow requires using the variant classification/Transcript Annotation functionality of each product. In SVS go to DNA-Seq > Variant Classification and set your analysis options making sure to select “Coding Variant Classification Report” from the Output Reports options and check the “Optional Filtering based on reports” with the Coding Variant Classification Report selected, leaving the default filtering selections on the next dialog.
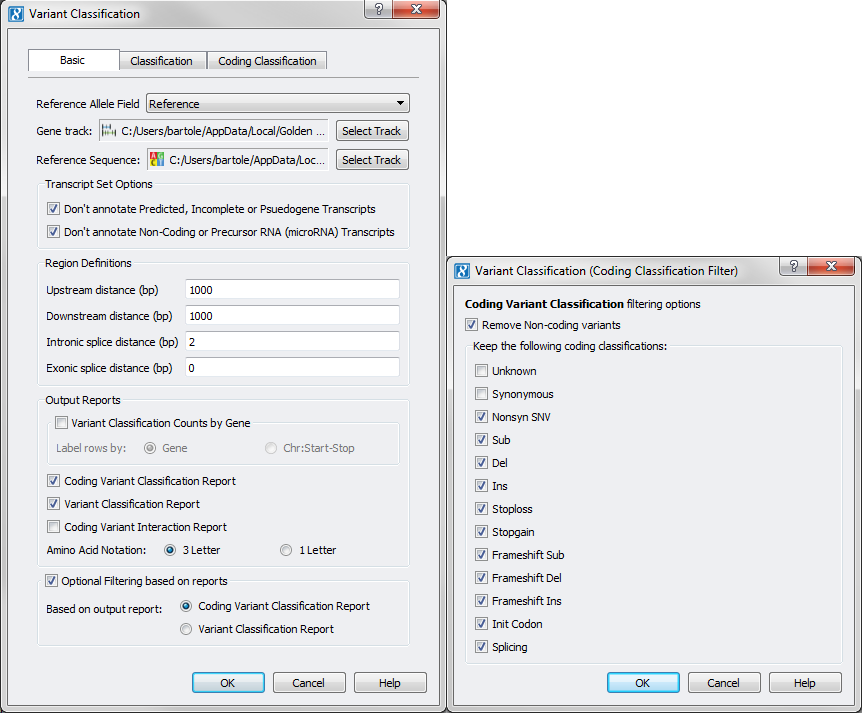
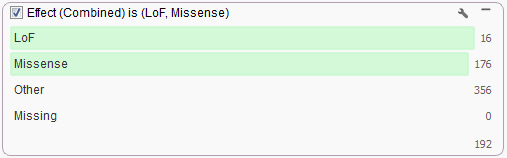
To obtain the results from Transcript Annotation within VarSeq all you need to do is annotate using a gene source. So, from your project go to Add > Annotation and select the “RefSeq Genes 105v2, NCBI” annotation source or your preferred gene source. Then create a filter card from the Effect column produced and select to keep only those variants classified as LoF or Missense. Results may vary slightly between the two products as the algorithms used for the analyses are slightly different. As a part of the future roadmap of SVS, we intend to eventually implement the Transcript Annotation algorithm from VarSeq to replace the existing Variant Classification functionality so results will be consistent between our two products.
This NGS data analysis workflow within VarSeq can be reproduced for new samples simply by going to File > Save Project as Template. Then the next time you load a new sample in a VarSeq project, simply select the custom template.
If you have any questions or you need help with your workflows in either SVS or VarSeq please email us at [email protected] and we will be happy to assist.