Huntington’s Disease (HD) Background
Huntington’s Disease (HD) is an autosomal dominant neurodegenerative disease that is caused by a mutation in the huntingtin (HTT) gene resulting in 36 or more CAG trinucleotide repeats in exon 1. Individuals affected by HD experience motor disorders including involuntary movements and poor coordination, cognitive impairments showing a decline in thinking and reasoning and psychiatric disorders resulting in changes in personality and mood. Typically, symptoms of HD occur between ages 30-40, but age of onset can occur as early as in teenage years or at any point throughout aging. Current research suggests that age of onset, as well as symptom severity, depends on the number of CAG repeats. As the number of CAG repeats increases, the severity of symptoms increases, and individuals are more likely to show signs of HD at an earlier age (1).
Genetic Counseling
It is important to mention that when the HTT gene is passed from one generation to the next, the length of CAG trinucleotide repeats increases, a phenomenon called anticipation. This is an important consideration for the variant under investigating because individuals who have 36-39 CAG trinucleotide repeats have an allele that has reduced penetrance and may not experience HD symptoms until very late in life if at all. In terms of genetic counseling, HTT variants meeting these criteria become very interesting to investigate as these individuals are at risk for having children that do develop the disorder (2,3).
Case Scenario
A 76-year-old man who is a retired welder presented with involuntary movements of the face and hands in his early 50s. He was evaluated by a neurologist and was diagnosed with “metal poisoning” and because symptoms were mild did not receive any treatment. Around age 71, he developed some paranoia believing he was being influenced by magnetic fields from having poisonous metals in his body. Additionally, his balance and involuntary movements worsened which lead to a workup including a 3-day EEG, but no abnormalities were found. The patient does not have any family history of involuntary movements or psychiatric disorders. Genetic testing revealed that he had 36 CAG repeats in one allele in the HTT gene and 17 repeats on the other allele. Treatment with risperidone resulted in improvement of chorea and paranoid delusions.
The patient above contained a variant within the HTT gene that produced 36 CAG repeats but still presented with HD symptoms in old age well after reproduction. In cases when HD symptoms are subtle, the possibility remains that those symptoms can be overlooked or thought to be typical symptoms of aging. This case emphasizes the interest and importance in investigating variants that result in 36-39 CAG repeats.
Variant Investigation
Due to the mendelian autosomal inheritance pattern of variants within HTT, VSClinical’s application of the ACMG guidelines will help us deep dive into an interesting variant associated with HD.
To begin, we can significantly narrow down the thousands of variants from our patients’ exome data based on quality alone. To filter on quality, we can use the genotype qualities field from our VCF file and set the threshold value to 50 to collect variants that have a genotype quality of greater than or equal to 50 (Figure 1).

Then we will want to look at variants that are specifically located in the huntingtin gene (HTT) because it is known that the CAG repeat region is located on Chromosome 4 within exon 1 of the HTT gene. We can do this by running the Match Genes List algorithm from the computed data list and entering in the HTT gene since it is our only gene of interest (Figure 2).
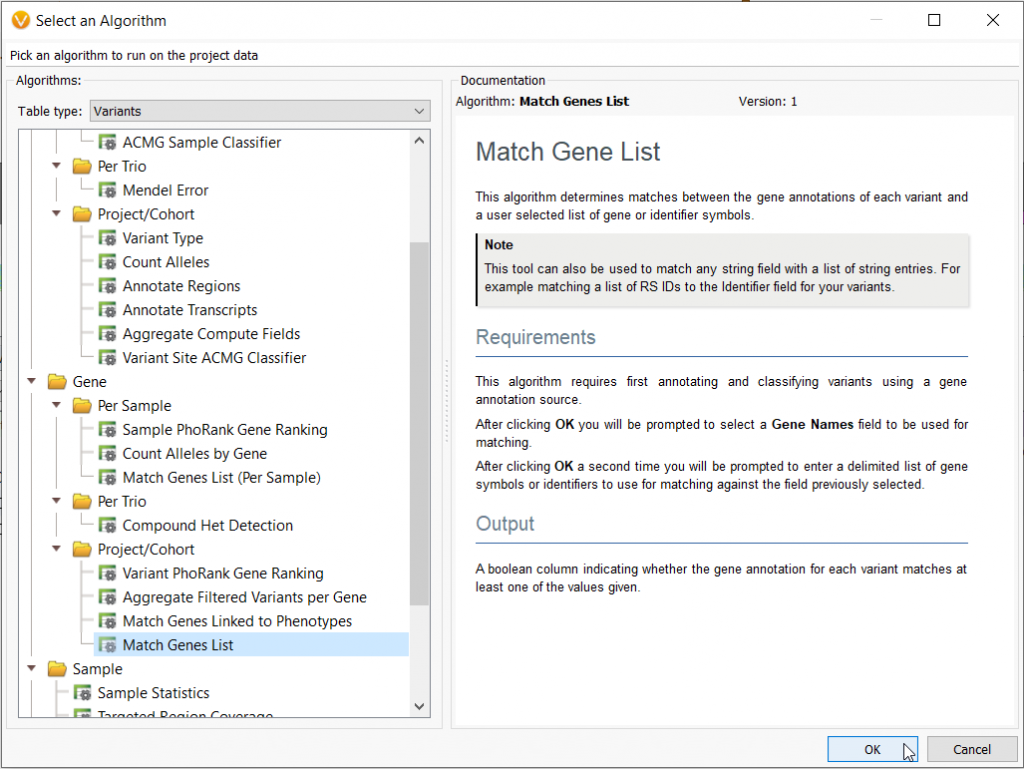
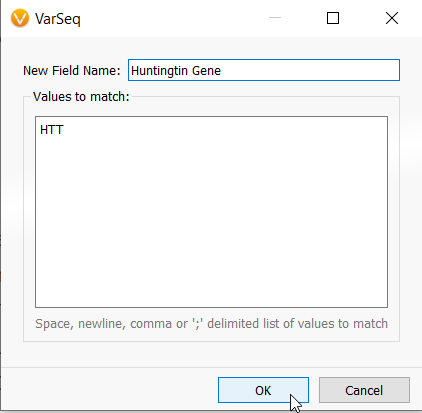
Figure 2: VarSeq can apply the Match Genes List algorithm (A) so the user can define a specific set of genes that harness a defined phenotype or disease. In this case, one gene related to Huntington’s Disease (B).
Finally, because we are looking for extra CAG repeats inserted into the gene, we can use the sequence ontology field from the RefSeq genes annotation source and isolate in-frame insertion variants (Figure 1). After applying all of the above filter logic, we successfully narrow our search to one variant on chromosome 4, within exon 1 of the HTT gene that results in a chain of 38 CAG repeats (Figure 3). We can now flag this variant for evaluation according to the ACMG Guidelines within (Figure 4)
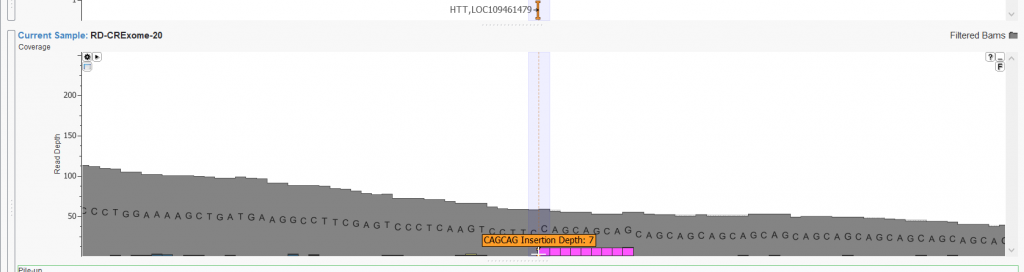
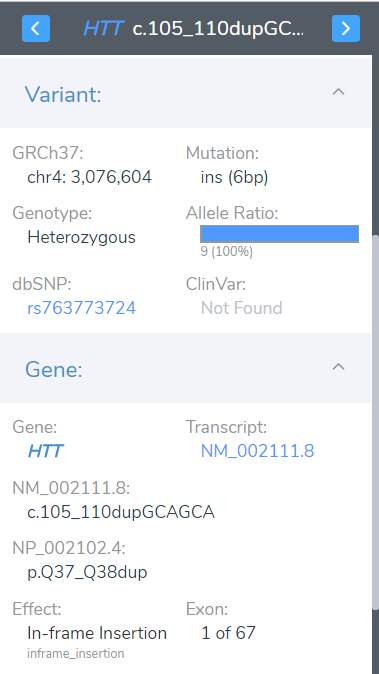
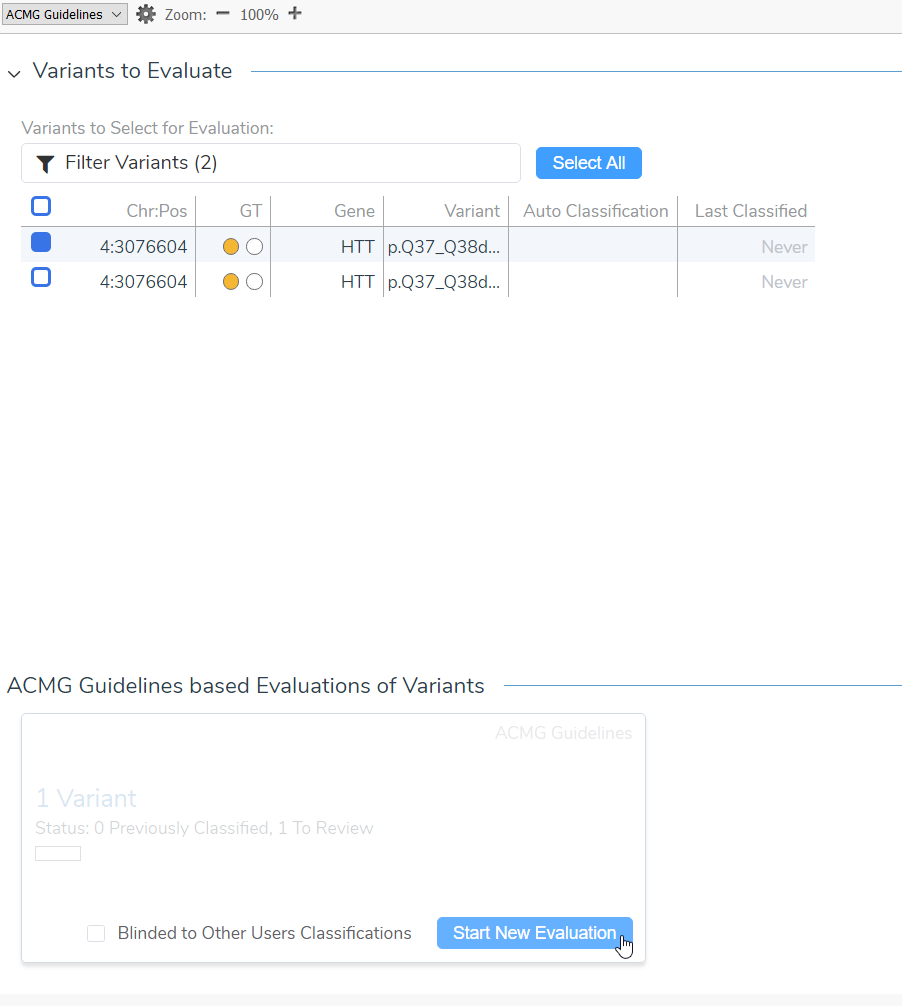
Figure 4: A In-frame insertion variant (c.105_110dupGCAGCA) (A) was opened in VSClinical to be evaluated according to the ACMG Guidelines (B)
ACMG Variant Classification
Once our variant is opened in VSClinical, we see that the HTT 105_110 variant has mixed recommendations with both benign and pathogenic scoring criteria that need to be addressed.
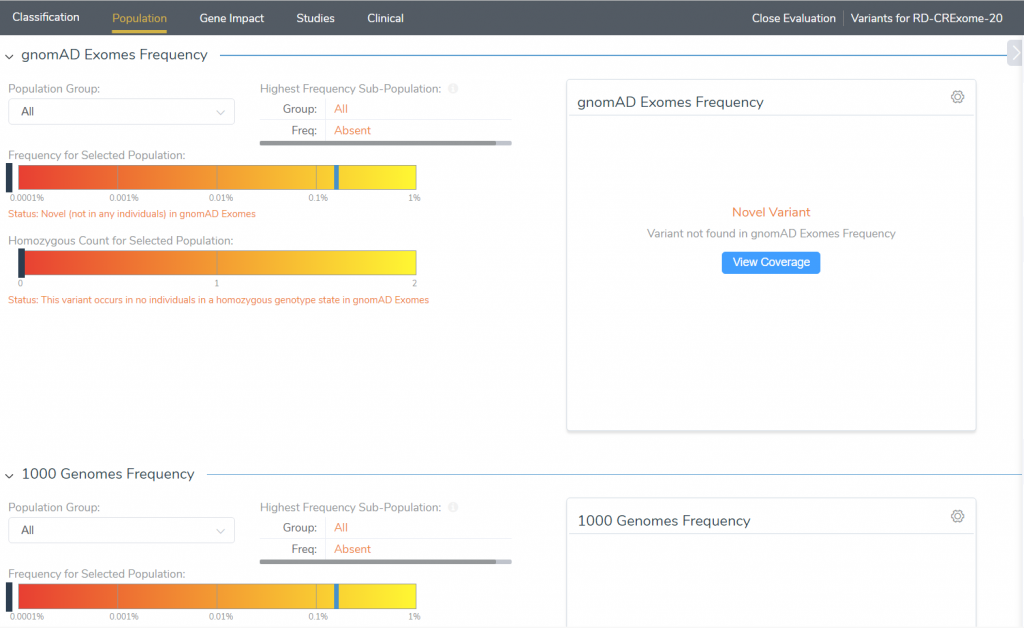
If we start our investigation with by evaluating the suggested pathogenic criteria, we can see that the variant is novel according to both gnomAD and 1000 Genomes frequency tracks enabling the inclusion of the moderate PM2 criteria into our classification (Figure 5).
Next, by analyzing the impact of this variant on the gene, we are recommended the inclusion of benign criteria BP3 as well as BP4. Interestingly, exon 1 of the HTT gene is a repeat region and individuals who do not have HD have roughly 20 CAG repeats. However, in Huntington’s disease, adding too many repeats in this region establishes presentation of Huntington’s disease symptoms. Additionally, PhyloP and GERP++ maintain that this region is not conserved. The CAG repeats in this region of exon 1 are normal, however, the number of repeats varies between individuals explaining why the computational evidence sustains the region is not conserved (Figure 6). In lieu of these caveats, it is important to review the studies tab within VSClinical before including the BP3/BP4 benign criteria.

After reviewing the studies tab, we can see other assessments wherein variants also with exon 1 involve the addition of several CAG repeat inclusions which are considered pathogenic according to ClinVar. Further investigation through searching google, google scholar and PubMed within VSClinical, elaborates on HD pathogenesis and the potential pathogenic risk for the HTT 105_110 variant or other variants that cause reduced penetrance (Figure 7). Due to functional in vivo studies and genetic testing of CAG repeats in HD individuals we can add a supporting pathogenic criterion of PS3 (Figure 8).
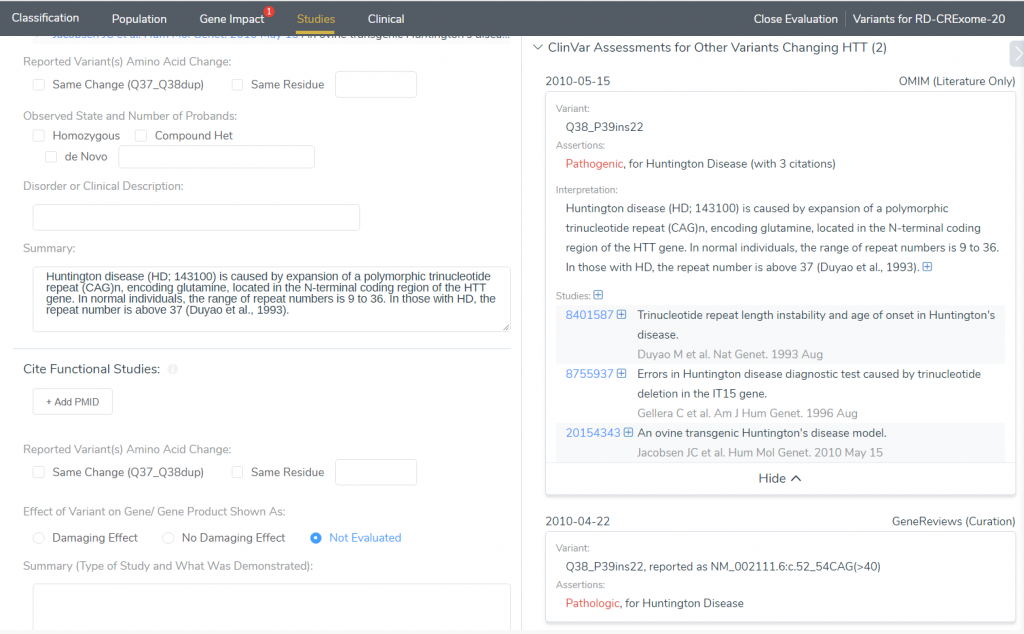
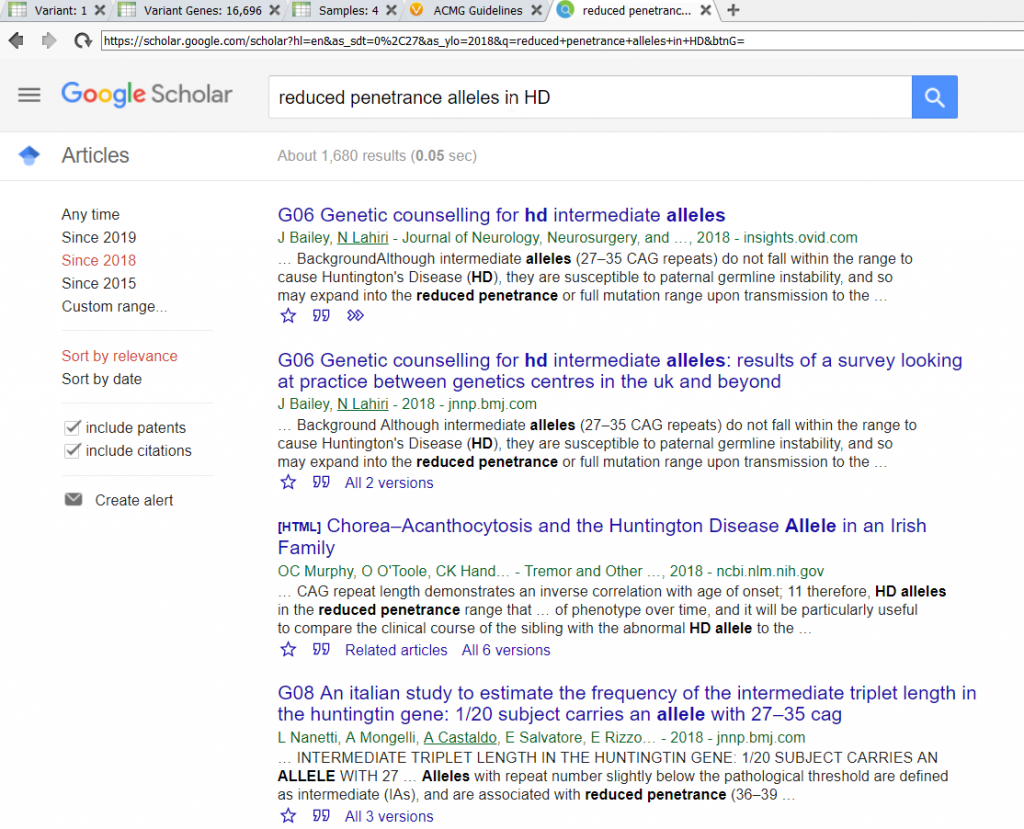
Now we can return to addressing the previous suggested benign criteria. There was not any evidence found suggesting the protein length changes as a result of CAG inclusions so we can accept the benign BP3 criteria. At this point, the ACMG classifier is suggesting a likely pathogenic classification for our variant. But, what about the computational evidence? If we accept the benign BP4 classification that this region is not conserved we rest on a classification of uncertain significance, which suits the controversial nature of HTT reduced penetrance variants of 36-39 CAG repeats within this region (Figure 9).
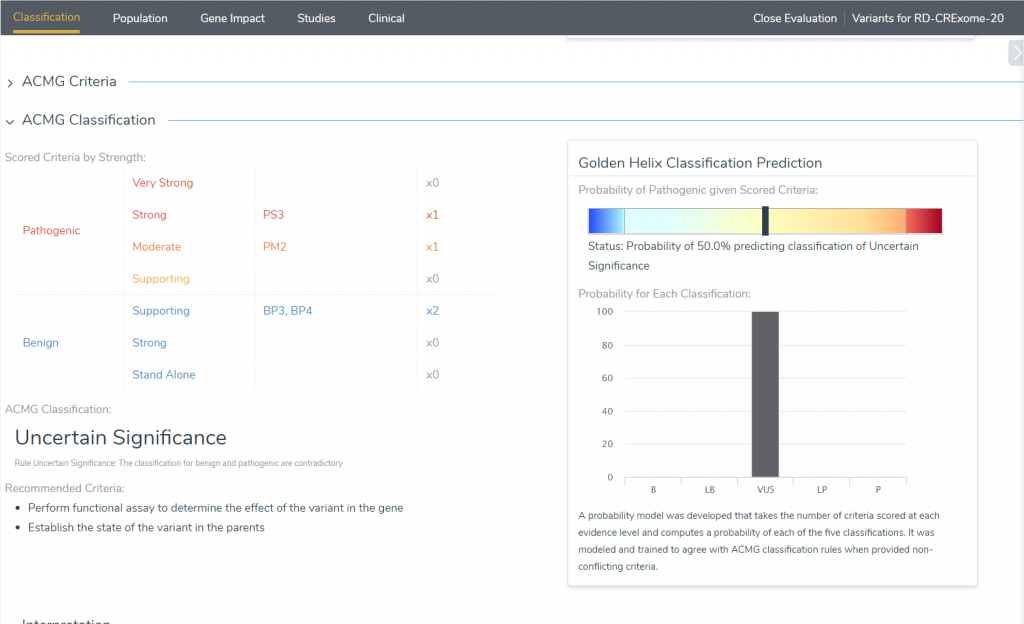
Conclusion and Summary
As all clinicians recognize, not all variants are so obviously benign or pathogenic. The application of research and previous studies is critical in analyzing the overall classification of some disorders. Huntington’s Disease exemplifies the nuanced nature of rare disease analysis, where reduced-penetrance variants require careful interpretation of both population data and functional evidence. Regarding the HTT 105_110 variant, it cannot confidently be defined as pathogenic and future studies and analyses will reveal the pathogenic risk associated with this variant. For genetic counselors, this variant is of great interest and the phenomenon of reduced penetrant alleles should not be disregarded. VSClinical was able to successfully interpret a variant with a complicated story, demonstrating how purpose-built variant interpretation software balances automated scoring with guided literature review. The automation of analyzing this variant through VSClinical helped us consider each criterion alone, but also prompted the utilization of literature review. In this edge-case variant is important to emphasize the manual component of variant analysis prompted by the automated workflow.

References
(1) Savitt D. et al. Clinical phenotype in carriers of intermediate alleles in the huntingtin gene. J of the Neurological Sciences. 2019; 402:57-61.
(2) Migliore S, Jankovic J, Squitieri F. Genetic Counseling in Huntington’s Disease: Potential New Challenges on Horizon?. Front Neurol. 2019;10:453.
(3) Kay C, Collins JA, Miedzybrodzka Z, et al. Huntington disease reduced penetrance alleles occur at high frequency in the general population. Neurology. 2016;87(3):282–288.
Check out some of the other blogs in this series:
- Variant Interpretation with VSClinical: Clinical Example for Congenital Indifference to Pain
- Variant Interpretation with VSClinical: Evaluation of an X-linked recessive mutation
- Variant Interpretation with VSClinical: Evaluation of Hypertrophic Cardiomyopathy
- Variant Interpretation with VSClinical: Congenital Myasthenic Syndromes (CMS)
- Variant Interpretation with VSClinical: Non-Small Cell Lung Cancer
- Variant Interpretation with VSClinical: RET-KIF5b Gene Fusion



