Secondary Analysis with Sentieon: Rapid and Accurate Variant Calling
This blog post will cover the utilization of secondary analysis tools to produce a list of high-quality variants and associated coverage data. This data will serve as the main, importable content for the tertiary stage of analysis where variants are interpreted and classified for their impact on a patients’ disorder. We will start by generally covering the process of where the secondary analysis begins.
Following the production of a sample FASTQs from any sequencer, Sentieon is the secondary analysis processing of aligning the reads to a reference assembly and ultimately variant calling. The more specific layout of this process is outlined here:
- Read Alignment: Align the random assortment of reads in the FASTQ against a reference sequence (FASTA) file and produce initial BAM file.
- Mark Duplications: Isolate and remove duplicate reads (optional to skip if working with amplicon data).
- Realign Insertions/Deletions: Quality improvement step to realign against known indel sites.
- Recalibrate Base Quality Scores: Quality improvement step to adjust for over or under-estimated base quality scores in sequence reads.
- Variant Calling: Process of determining active regions of the genome, and with consideration for haplotype likelihoods to ultimately assign the genotype and produce a list of variants in the VCF file.
This general process can be done for both germline and somatic variant calling, all listed and described on the Sentieon site.
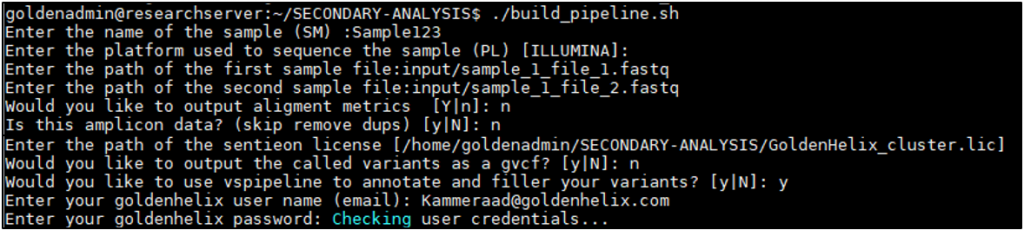
Using a direct example, Sentieon was previously installed and a batch of sample FASTQ files was stored in a preferred input directory. To simplify the command line interface with Sentieon, an automated build pipeline script is shipped with the Sentieon installation directories. This build pipeline script is initially designed for a single sample. However, the Golden Helix Support Team can assist users with developing a batch script to run multiple samples sequentially. To use the build pipeline script, the user will first move to the secondary analysis directory available after Sentieon installation.
Basic single sample instructions are as follows:
| STEP | COMMAND |
| Change to Secondary Analysis directory | cd SECONDARY – ANALYSIS |
| Build script to process samples | ./build_pipeline |
| Run call variant script | bash output_sample/call_variant |
Running the build_pipeline script will guide the user through the process of:
- Designating the sample name: Sample123
- Sequencing platform used: Illumina
- Assign FASTQ input directory/file paths: input/sample_1_file_1.fastq
- Same process for second FASTQ: input/sample_1_file_2.fastq
- Option for output alignment metrics: y or n
- Optional deduping: n or no to amplicon data
- Path to Sentieon license file:
/home/goldenadmin/SECONDARY-ANALYSIS/GoldenHelix_cluster.lic - Option for gvcf output: y or n
- Option for running VSPipeline: y or n with login
Once the build pipeline is complete, an output directory will be made that matches the assigned sample name. In the output directory will be a “call variants script” that will run the algorithms for the alignment and variant calling process described in the outline above. Run this script with the command bash output_sample/call_variants:

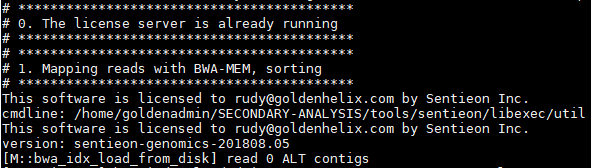
Once the scripts start, you’ll see the algorithm details run in the terminal. Starting with alignment to produce the BAM:
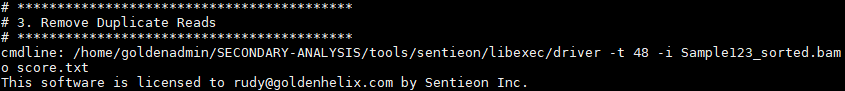
Then marking and removing duplicate reads:
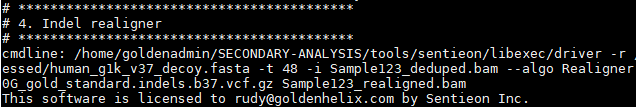
Followed by indel realignment and base quality score recalibration:

And finally calling the variants to produce the VCF file:
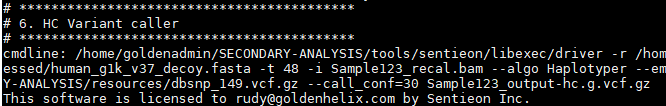
Batch Script for Multiple Samples
Similarly, a batch script can be used to run multiple samples. The modifications for the batch run will be integrated into the call_variant.sh script. Essentially, the same process mentioned above will run through all samples which can be set up to process a complete sequencing run in one go. This is an obvious desire for any high throughput lab handling a massive sample load on a regular basis. Our “Automating NGS Gene Panel Analysis Workflows with Golden Helix” webcast showcases this automated capability for larger-scale NGS analysis workflows.
VSPipeline
The final stage of this secondary process will be utilizing a premade project template to automate the variant filtering and variant prioritization. Filtering criteria may include quality fields from the VCF, selecting out commonly known benign variants in clinical databases, or selection to capture variants in genes associated with targeted phenotypes. The saved project template will be selected in VSPipeline to batch load all samples with called variants and process those variants through the filter workflow. The filtered set of variants can then be automatically exported, or explored in the project for the investigation of final interpretation and classification. The next part of our blog series will be covering how VSPipeline is deployed and what optional outputs can be generated.
We understand that each users’ methodology or workflow design can vary. It is our goal to assist with installation and successful deployment of these tools tailored for your desired pipeline. If interested in learning more about the secondary analysis solution with Sentieon, please contact [email protected] with any of your questions or concerns.
Next in this series is “Automating & Standardizing Your NGS Workflow: Part II” explaining the initial steps of tertiary analysis with VSPipeline.



