In this blog post, I am very excited to talk about The Broad Institute’s release of the latest version of gnomAD, v 3.1.2, which is now available for use as an annotation source in your SVS or VarSeq projects. For VarSeq users, I also want to point out that gnomAD v3.1.2 can also be used as a population frequency in VSClinical! You can add gnomAD 3.1.2 to your VarSeq projects by going to the Add Icon > Variant Annotation > then begin to type gnomAD into the annotation search bar. If gnomAD Genomes 2.2.1 was already incorporated into your project you can update to the new version within the project by clicking on the blue update icon in the top right-hand corner.
What are some of the main changes between gnomAD v2.1.1 and gnomAD 3.1.2?
- A 5-fold increase in the number of whole genomes, thus increasing the analysis power for those interested in non-coding regions
- Over 3,000 new samples were added to increase the ancestral diversity of the database.
- There is now a designated population label for samples of Middle Eastern and Amish ancestry.
- Roughly 489,619,768 more variants (passing quality filters) were added.
- Better rsID matching using both locus and allele information instead of just chromosomal locus only.
- Fixed homozygous alternate allele depletion adjustment for samples with heterozygous non-reference genotypes.
At a more detailed level, the sample QC was improved between gnomAD v2.1 and v3. Namely, 1) the use of normalized coverage for both X and Y to infer sample sex and 2) a new way of filtering outliers based on QC metrics.
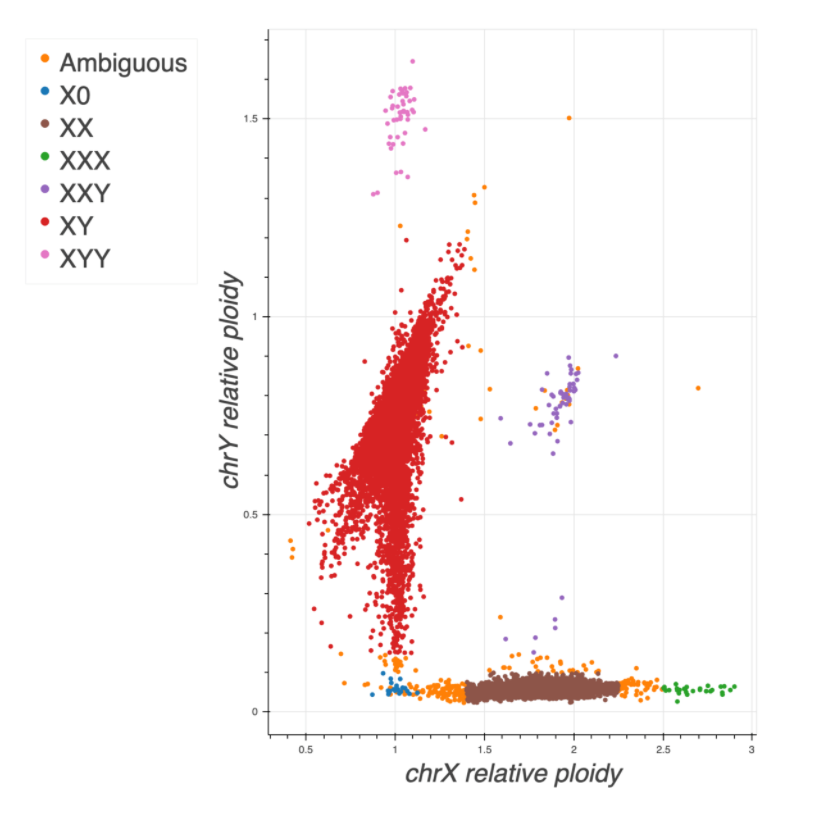
- To infer the sex of a sample, mean coverage was computed on non-PAR regions of the X and Y chromosomes. These values were normalized using the mean coverage on chromosome 20. Using Hail impute-sex function, an inbreeding coefficient F was computed (Figure 2).
- A new strategy was used to filter outlier samples based on QC metrics. Previously, samples were grouped based on ancestry, but in gnomAD v3, the Broad Institute team regressed out the principal components computed during ancestry assignment and filtered the samples based on the residuals for each of the QC metrics from the Hail sample_qc module.

Golden Helix was very excited to receive and curate this database for our customers to use in their projects. gnomAD Genomes v3.1.2 is currently available for both GRCh 37 and 38 genome assemblies. If you would like to explore more details about the latest gnomAD release, please feel free to read through the gnomad news page for the gnomad v3.1.2 release.



