
At Golden Helix, we’ve supported the HGVS variant nomenclature standard since the very first release of VarSeq. The HGVS standard provides a consistent and precise language for describing sequence variants in the context of gene transcripts and is a critical part of clinical genomics workflows.
As with all living standards, HGVS continues to evolve, refining how we describe increasingly complex genomic events. Along the way, we’ve kept pace by extending VarSeq’s Annotate Transcripts algorithm to handle new edge cases and provide users with fine-grained control over how their HGVS annotations are computed and displayed.
Some users adopt the latest recommendations from HGVS, while others prefer more stable community practices aligned with clinical reporting pipelines. We support both of these approaches, by offering customizable options that strike the right balance for your workflow. Today, we’re excited to announce support for one of the more recent additions to the HGVS standard: repeat notation.
Why Repeat Notation Matters
During NGS analysis it is common to encounter variants that occur in repeat regions where a short nucleotide motif is repeated multiple times. Previously, deletions or duplications in these repeat regions could only be annotated using standard HGVS notation without regard for the surrounding repeated sequence. This can be problematic when analyzing clinically significant short tandem repeats (STRs).
A compelling example is the HTT gene, where repeat variation has direct clinical implications for Huntington disease. The HTT gene contains a repeated motif in exon 1, and the number of repeats in this region varies significantly across the population. Importantly, the number of repeats in the reference sequence is somewhat arbitrary. While the GRCh38 reference sequence contains 19 copies of the repeat motif, in the general population, individuals may have anywhere between 6 and 26 copies, which are considered benign. Pathogenic expansions typically involve more than 40 repeats.
Consider the variant 4:3074877 -/CAGCAGCAGCAG shown below:
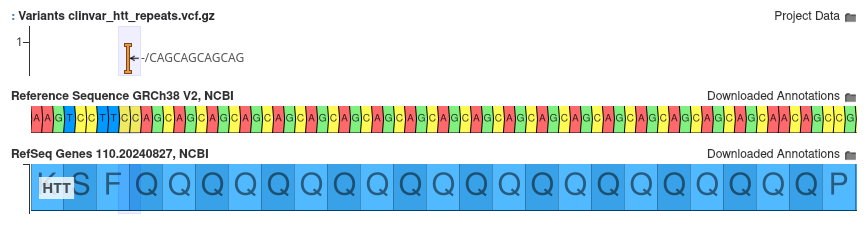
This variant occurs in the context of the HTT repeat region, with the variant inserting an additional four copies of the repeated motif. Using the default notation, this variant would be annotated as c.110_111insGCAGCAGCAGCA. This notation indicates an insertion of 12 nucleotides from positions 110 to 111, but it fails to tell us anything about how many total copies of the motif are present in the sample.
Using the new repeat notation feature, VarSeq will instead annotate this variant as c.54GCA[23]. This alternate representation tells us that, at position 54, the clinically relevant repeat motif “GCA” occurs exactly 23 times in the sample. This representation makes it instantly clear that the sample falls within the benign range, providing more clinically relevant information than the traditional insertion notation.
Syntax of HGVS Repeat Notation
The HGVS repeat notation follows the syntax structure shown below:
prefix + position_repeat_start + repeat_sequence + [ copy_number ]
Where:
- prefix defines the transcript and notation type (e.g.,
NM_006172.4:c.) - position_repeat_start indicates the starting position of the repeat
- repeat_sequence is the actual nucleotide sequence that constitutes the repeat unit
- copy_number is the number of times the repeat motif is present in the sample
This standardized format ensures consistency across different analysis workflows while providing clear, interpretable information about repeat-altering variants.
How to Use This Feature in VarSeq
To take advantage of this new feature:
- From VarSeq select Add → Computed Data
- Under the Project/Cohort folder, Select the Annotate Transcripts algorithm
- Select your desired gene track
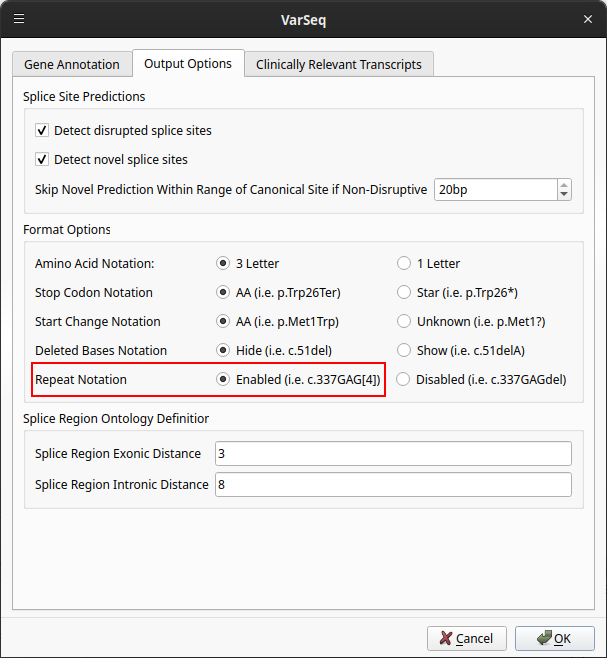
- Open the Output Options tab
- Click the Enabled option next to Repeat Notation
Conclusion
This new feature reflects our ongoing commitment to supporting high-quality, standards-compliant transcript annotation. Whether you’re analyzing exomes, panels, or whole genomes, VarSeq’s support for flexible and accurate HGVS repeat notation empowers you to communicate results with greater precision. If you’re interested in learning more about VarSeq’s powerful annotation capabilities, please reach out to our team.



