Streamlining the ACMG Guidelines and Providing Scoring Recommendations
As we discussed in our recent webcast on VSClinical, the process of scoring the ACMG guidelines requires evaluating evidence for the connection between a variant and the disorder or condition being evaluated by the genetic test for an individual. These lines of evidence cover clinical presentation, gene function, bioinformatic annotations and in-silico predictions as well as population frequencies. Some of ACMG scoring guidelines that rely on evidence that can be derived through bioinformatic methods can have their status automatically computed with supporting evidence documented.
Auto Scoring and Previous Interpretations as part of the VSClinical workflow
Core the ability to streamline any type of ACMG guideline analysis is having a vast repository of genomic annotations and variant annotation algorithms. VSClinical leverages the years of innovation and iterative development we have put into VarSeq to build the most advanced and complete annotation engines in the industry. For the 18 criteria of the 33 in the ACMG guidelines that we streamline, we are relying on 7 annotation sources and annotation algorithms:
- Variant Interaction with RefSeq Genes, NCBI: Our gene annotation algorithm provides us with the sequence ontology of a variant against a transcript, as well as the selection of the default transcript and details about how far along we are in the coding sequence of the gene.
- Splice site predictions: Our implementation of four splice site prediction algorithms allows for the detection of splice disrupting variants around canonical splice sites.
- gnomAD and 1000 Genomes population frequencies: Our curation of these sources carefully tracks not just the allele frequencies, but also the number of homozygous and hemizygous genotypes are present in individuals (a criteria for considering a variant benign).
- ExAC Gene Constraint Scores: Containing estimates at a gene level of whether certain types of variants occur less often than expected (indicating selection against that type of variant) in the large ExAC exome cohort.
- OMIM: Our gene and phenotype curation of OMIM allows for certain aspects of the analysis to be aware of whether the gene has been associated exclusively with dominant model disorders.
- Functional Prediction: computed on a transcript-aware basis on the 100-way multiple sequence alignment, our SIFT/PolyPhen2 missense predictions provides supporting computational evidence for pathogenic and benign scoring critera.
- Conservation Scores: also running on the 100-way MSA, the two complementary conservation scores from our implementation of GERP++ and phyloP providing supporting computation evidence.
These annotations along with the presence of the current variant in the database of previous variant classifications using VSClincial are used to provide a list of auto-scored ACMG criteria and a final classification for every variant in your project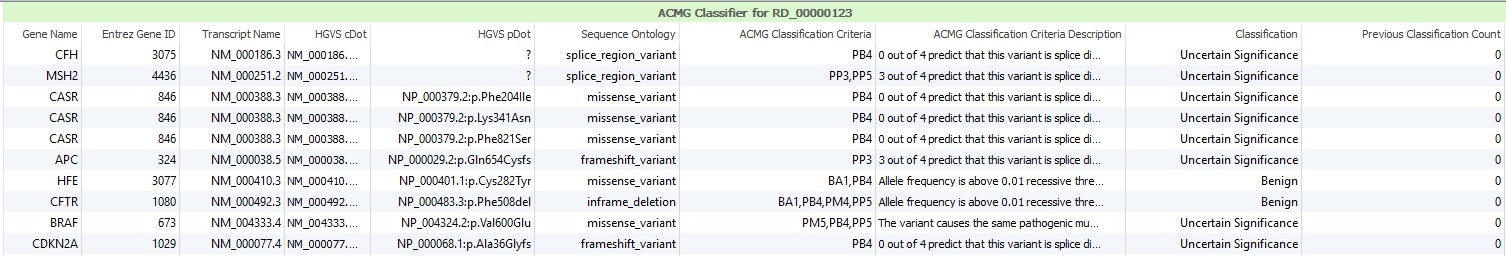
Recommendations and Auto-Interpretations
Once evaluating variants in VSClinical, the auto-scored criteria are provided as recommendations in the very first screen, but it is up to the user to decide whether to accept each recommendation. Along with the suggestions are helpful descriptions of the evidence that supports the recommendation. The links on each criteria box jumps to the tab and the section containing the contextual evidence related to each criteria.
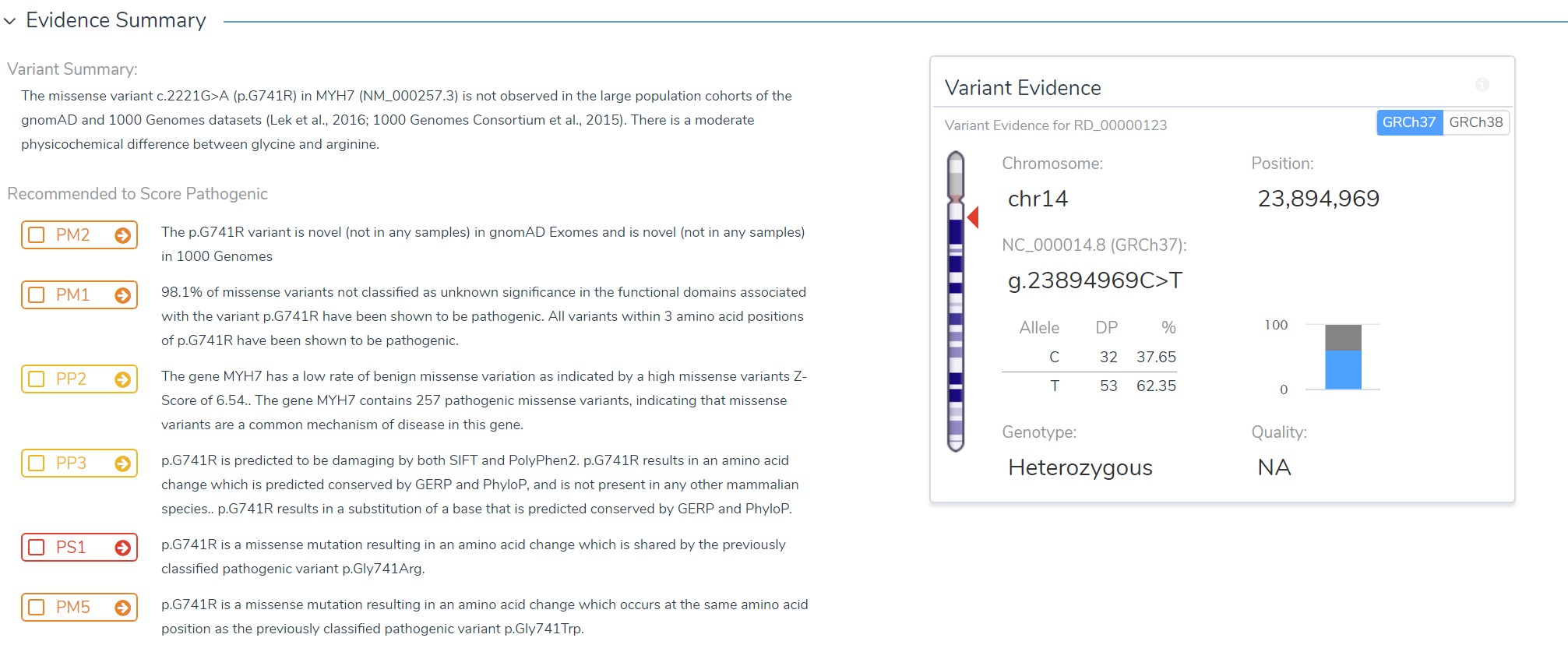
This variant has quite a few recommendations for pathogenicity (it is a well-established causal mutation for cardiovascular disease).
Some questions in the ACMG guidelines are really compound in their nature and require a couple supporting pieces of evidence to meet the threshold for their scoring. VSClinical breaks these questions out into answerable statements and provides recommendations for each one individually.
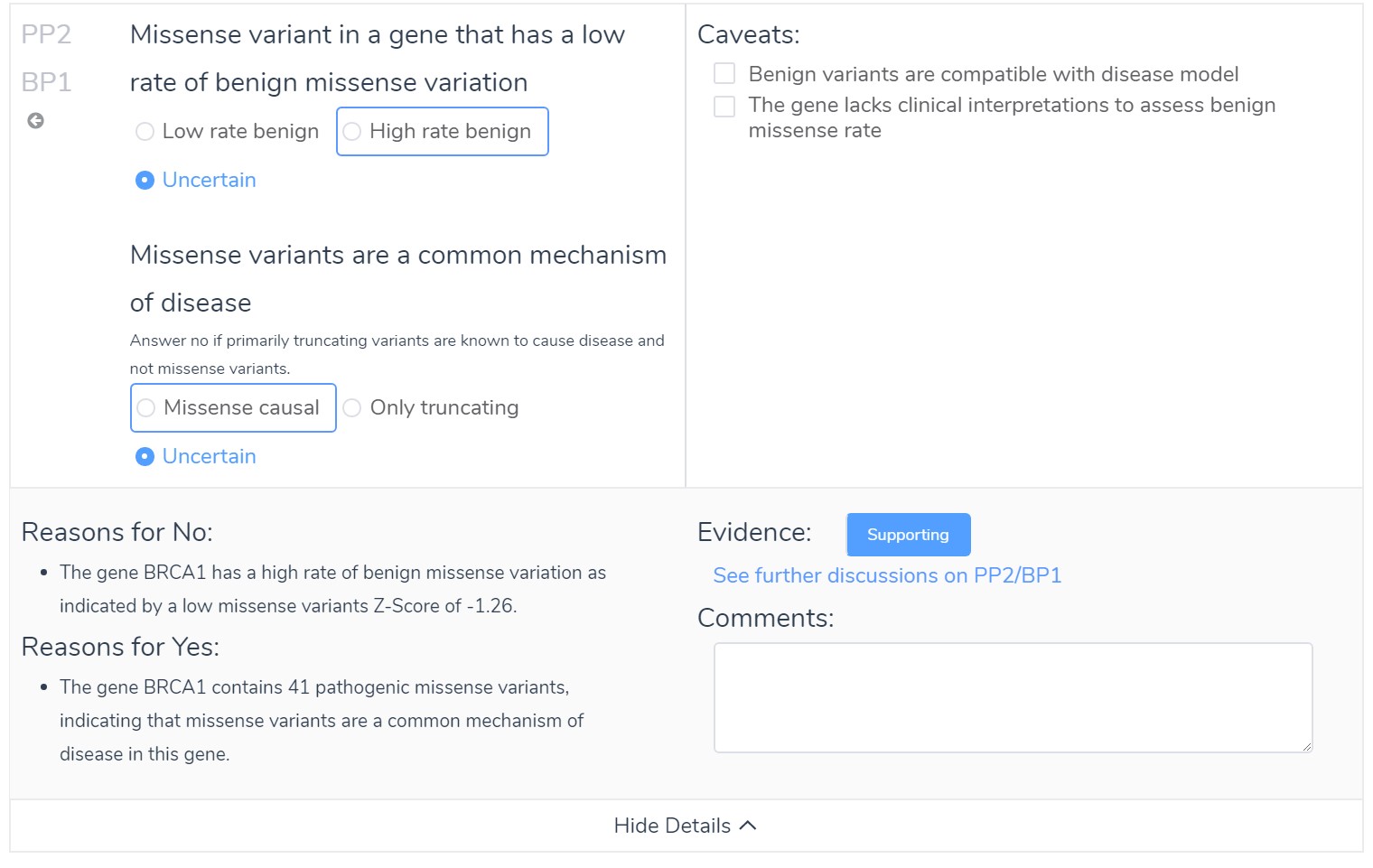
For BRCA1 we have evidence that there is a high rate of benign missense variation, but the high number of pathogenic missense variants demonstrates that missense variants mare a common mechanism for disease.
Dealing with Conflicting Criteria
Sometimes the evidence does not line up cleanly in one column or another when it comes to classifying variants as pathogenic or benign. For these situations, we have the Golden Helix Classification Prediction which uses a trained probability model to look at the scored criteria you have completed and quantify the probability for each of the five classification states as well as total pathogenicity prediction.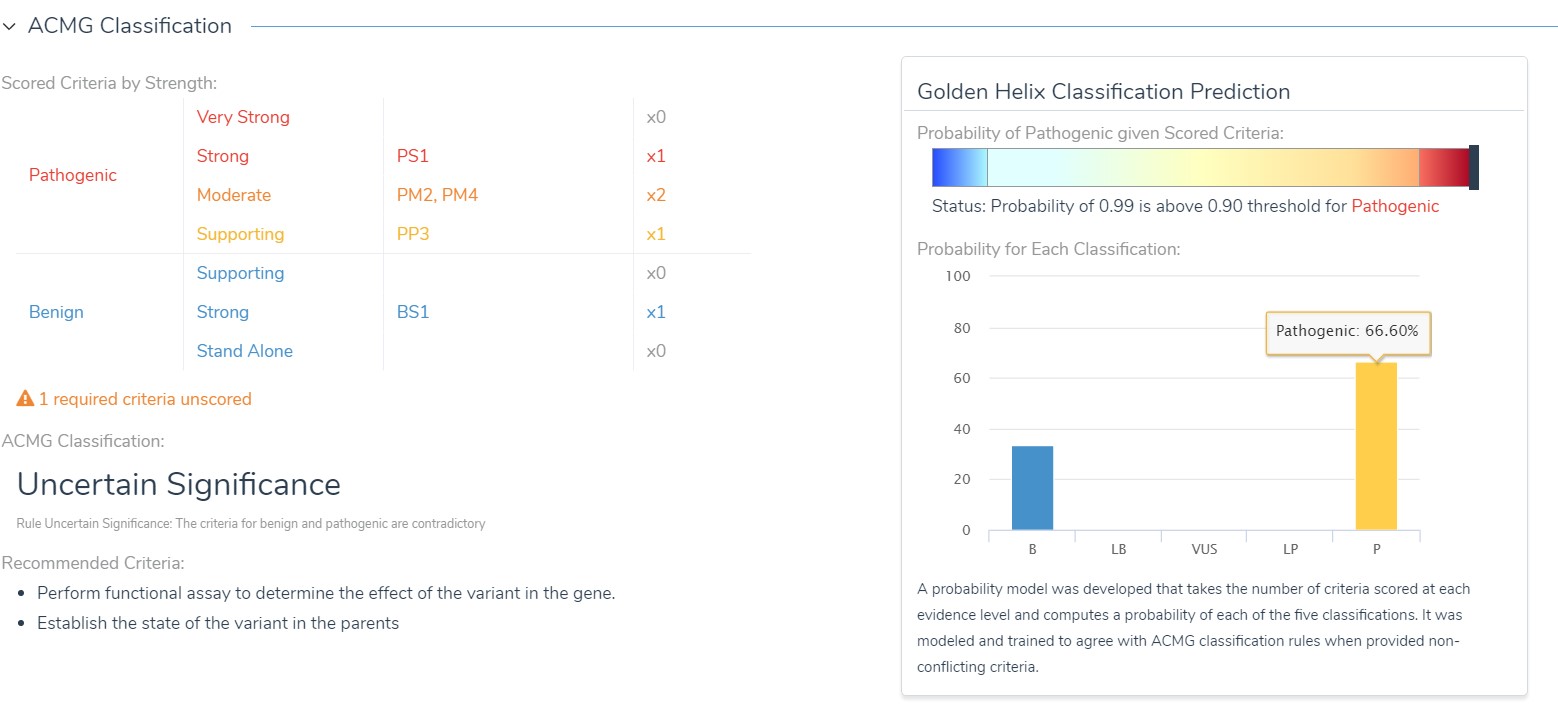
In this case, we may feel confident enough in our evidence to classify this variant as Pathogenic, even though we have the strong benign criteria BS1 scored for this variant because it occurs in a higher than expected frequency for the disorder in large cohort populations.