Functional Predictions and Conservation Scores in VSClinical
Several algorithms have been developed to predict the impact of amino acid substitutions on protein function and quantify conservation of nucleotide positions. These methods provide vital supporting evidence to clinicians when interpreting variants in accordance with the ACMG guidelines. The two most popular functional prediction algorithms are SIFT and PolyPhen2, while the most common conservation score metrics are GERP++ and PhyloP LRT.
SIFT was developed in 2001 by Ng and Henikoff and utilizes multi alignment information to classify substitutions as damaging or tolerated [1]. For a given protein sequence alignment, SIFT constructs a position-specific scoring matrix (PSSM), which encodes the probability of each amino acid at each position of the protein. These probabilities are computed by averaging the weighted observed amino acid frequencies with a set of unobserved frequencies computed using a Dirichlet mixture model [2]. Variants are classified as deleterious or tolerated based on set probability thresholds.
Polyphen2 is the creation of Adzhubei et al. and uses a Naïve Bayes Classifier to estimate the probability that a given substitution is tolerated or damaging [3]. Like SIFT, this approach utilizes a sequence weighted score, which incorporates pseudocounts. Specifically, PolyPhen2 uses the PSIC score to weight sequences and quantify conservation [4]. This score is combined with nine other metrics, including change in amino acid volume, congruency of the allele to the multiple alignment, and whether the mutation is a transition or a transversion. Some of these metrics are pulled from various database, which require significant storage overhead. In our own implementation we removed four of the metrics: the presence of the mutation within Pfam, the crystallographic beta factor, the normalized accessible surface area, and the change in accessible surface area. We found that removing these metrics had a negligible effect on performance while significantly reducing the algorithm’s storage requirements.
Developed by Davydov et al. in 2010, GERP++ uses a multiple sequence alignment in conjunction with a phylogenetic tree model to quantify conservation at a given position as the fraction of neutral substitutions rejected by evolutionary selection [5]. The algorithm uses dynamic programming to compute the scaling parameter that maximizes the probability of the alignment at a given position. This scaling factor quantifies the site’s rate of evolution relative to neutrality.
In 2010 Pollard et al. developed four statistical, phylogenetic tests for detecting departures from the neutral rate of substitution: a likelihood ratio test (LRT), a test based on exact distributions of numbers of substitutions, a score test, and a genomic evolutionary rate profiling test [6]. These tests were made available through the PhyloP software. Of these tests, the most commonly used is the PhyloP LRT, in which null and alternative phylogenetic tree models are fitted to an alignment through maximum likelihood estimation, and two times the difference of their log likelihood values is used as a measure of conservation.
Implementing and Comparing Functional Prediction Methods
We have developed our own implementation of these methods which can be utilized within VSClinical to provide computational evidence of pathogenicity. To compute these scores, we utilized UCSC’s multiple alignments of 100 vertebrate species [7]. In the case of SIFT and PolyPhen2, this results in a significant change to the nature of the algorithms’ input data, as both SIFT and PolyPhen2 utilize alignments queried using PSI-BLAST from a protein sequence database such as SWISS-PROT.
To establish the validity of our SIFT and PolyPhen2 implementations when run using the UCSC alignment data, we compared the performance of our implementations to the precomputed scores provided by dbNSFP [8]. For testing data, we used all ClinVar variants, with one or more stars, that were classified as either Pathogenic or Benign [9]. The results in Table 1 show the rate of correctly classified Benign and Pathogenic variants for our implementation, compared to the precomputed scores.
Table 1: SIFT and PolyPhen2 Comparison on ClinVar
| Accuracy | SIFT (Vertebrates) | PolyPhen2 (Vertebrates) | SIFT (dbNSFP) | PolyPhen2 (dbNSFP) |
| Pathogenic | 94.40% | 93.80% | 90.33% | 88.95% |
| Benign | 59.10% | 66.80% | 66.65% | 68.79% |
In both cases, our implementations outperform the precomputed scores in terms of accuracy on Pathogenic variants, while underperforming slightly in terms of accuracy on Benign variants. These results indicate that the use of UCSC’s species alignment data does not degrade the validity of the SIFT and PolyPhen2.
We also compared our implementations to several other precomputed functional prediction scores, including, MutationTaster, MutationAccessor, FATHMM, and Provean. These results are shown Table 2.
Table 2: Comparison to Other Functional Prediction Methods
| Accuracy | SIFT (Vertebrates) | PolyPhen2 (Vertebrates) | MutationTaster | MutationAccessor | FATHMM | Provean |
| Pathogenic | 94.40% | 93.80% | 84.72% | 80.84% | 95.41% | 85.96% |
| Benign | 59.10% | 66.80% | 58.59% | 60.65% | 74.00% | 77.90% |
Interestingly, FATHMM is the only algorithm which seems to unequivocally outperform our implementations of SIFT and PolyPhen2, both of which have exceptional classification accuracy for pathogenic variants when compared to the other methods. In terms of benign classification accuracy, SIFT and PolyPhen2 are competitive with both MutationTaster and MutationAccessor, while being outperformed by FATHMM and Provean. These results demonstrate that SIFT and PolyPhen2 continue to be competitive algorithms for the classification of missense variants.
Computational Evidence in VSClinical
In the upcoming weeks we will be putting the finishing touches on VSClinical, a tool designed to streamline the process of evaluating variants based on the criteria of the ACMG guidelines. This requires leveraging all available computational evidence for pathogenicity. When evaluating the gene impact of a variant, VSClinical will provide the clinician will all computational evidence associated with the variant, including splice site predictions, functional predictions, and conservation scores, and the relevant ACMG criteria will be automatically recommended based on the values of these scores.
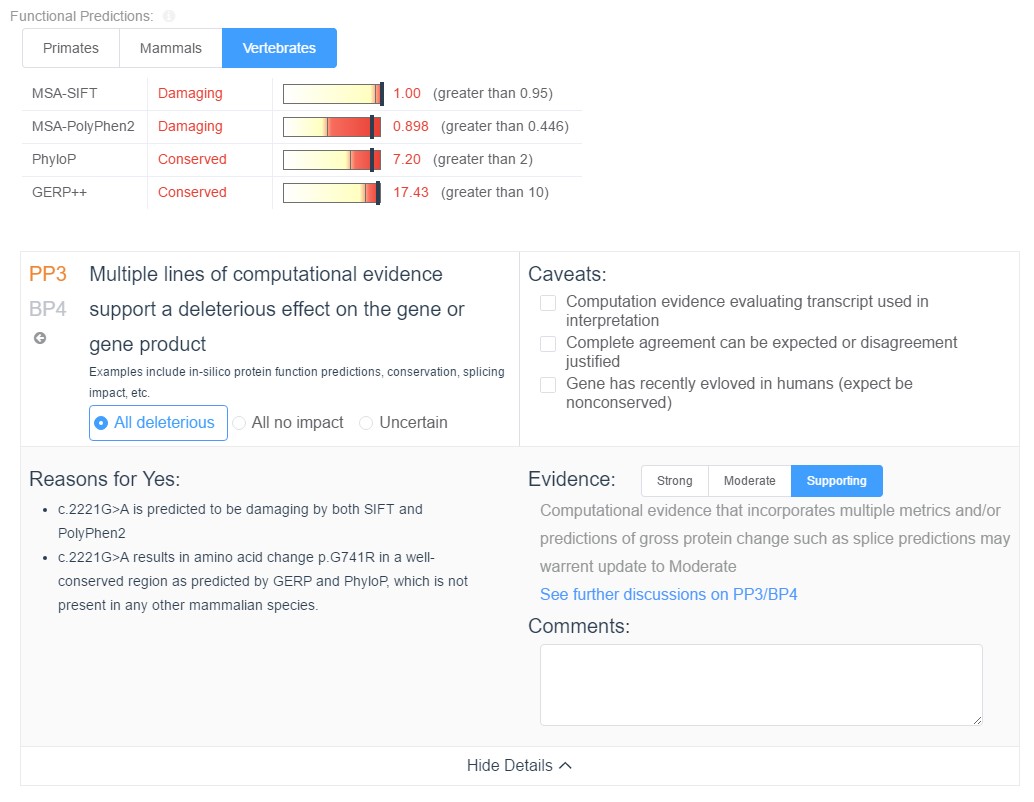
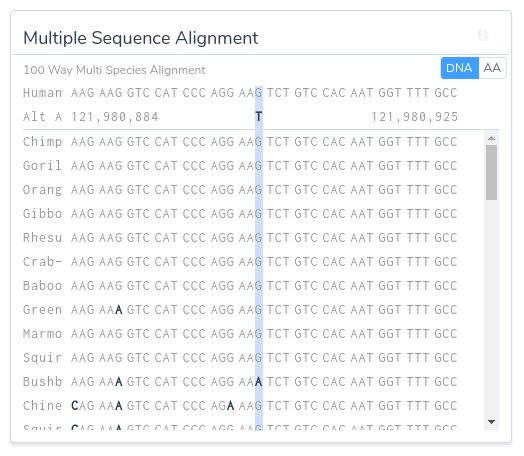
Additionally, all computed scores can be reevaluated on the fly in the context of any of the provided species groups: primates, mammals, or vertebrates. Along with the computational evidence, VSClinical also provides a convenient visualization of the multi sequence alignment for the surrounding region across the selected species, allowing clinicians to easily visualize the conservation status of the variant’s genomic region.
With these algorithms and tools, VSClinical provides the clinician with everything they need to evaluate the computational evidence for a given variant classification. If you have any questions on VSClinical or would like to see a personalized demonstration of the product to see how it works, please contact us by emailing [email protected]!
References
| [1] | P. C. Ng and S. Henikoff, “Predicting deleterious amino acid substitutions,” Genome research, vol. 11, no. 5, pp. 863-874, 2001. |
| [2] | S. Henikoff and J. G. Henikoff, “Position-based sequence weights,” Journal of molecular biology, vol. 243, no. 4, pp. 574-578, 1994. |
| [3] | I. A. Adzhubei, S. Schmidt, L. Peshkin, V. E. Ramensky, A. Gerasimova, P. Bork, A. S. Kondrashov and S. R. Sunyaev, “A method and server for predicting damaging missense mutations,” Nature methods, vol. 7, no. 4, p. 248, 2010. |
| [4] | S. R. Sunyaev, F. Eisenhaber, I. V. Rodchenkov, B. Eisenhaber, V. G. Tumanyan and E. N. Kuznetsov, “PSIC: profile extraction from sequence alignments with position-specific counts of independent observations,” Protein engineering, vol. 12, no. 5, pp. 387-394, 1999. |
| [5] | E. V. Davydov, D. L. Goode, M. Sirota, G. M. Cooper, A. Sidow and S. Batzoglou, “Identifying a high fraction of the human genome to be under selective constraint using GERP++,” PLoS computational biology, vol. 6, no. 12, p. e1001025, 2010. |
| [6] | K. S. Pollard, M. J. Hubisz, K. R. Rosenbloom and A. Siepel, “Detection of nonneutral substitution rates on mammalian phylogenies,” Genome research, vol. 20, no. 1, pp. 110-121, 2010. |
| [7] | University of California Santa Cruz, “Multiz 100 Way,” 2016 10 12. [Online]. Available: http://hgdownload.soe.ucsc.edu/goldenPath/hg19/multiz100way/. [Accessed 1 5 2018]. |
| [8] | X. Liu, C. Wu, C. Li and E. Boerwinkle, “dbNSFP v3.0: A one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs,” Human mutation, vol. 37, no. 3, pp. 235-241, 2016. |
| [9] | M. J. Landrum, J. M. Lee, M. Benson, G. Brown, C. Chao, S. Chitipiralla, B. Gu, J. Hart, D. Hoffman, J. Hoover and others, “ClinVar: public archive of interpretations of clinically relevant variants,” Nucleic acids research, vol. 44, no. D1, pp. D862-D868, 2015. |