In the world of genomics shaping precision medicine in oncology, the limiting factor is the time-to-sign-out of a fully interpreted molecular profile report. There are many components of the entire testing process that add to the turn-around time of each test. Many of these steps, such as sample prep, sequencing, and automated secondary analysis, are bounded and consistent in their time requirements. The hands-on time of the remaining analysis poses the highest risk and variability. The solution to control and master this step is specialized workflow software that supports efficient quality analysis, maximizes re-use of previous interpretations, and standardizes the process for authoring and signing out clinical reports.
In this blog series, I will be discussing VSClinical’s workflow for interpreting and reporting genomic mutations in cancer following the AMP guidelines. I will also be covering each of these discrete, hands-on analysis steps with a focus on efficiency and producing consistent high-quality results.
The Analysis Workflow for NGS Targeted Panels in Cancer
Let’s first review the hands-on analysis steps involved in creating a high-quality clinical report for targeted Next Generation Sequencing (NGS) assays.
- Quality Analysis: Ensure that a sample does not need to be re-run, that critical genomic regions are well covered, and that the variants are selected for hands-on interpretation are not false-positives.
- Patient Mutation Profile: Inputting the small variants, CNVs, and gene fusions detected by the genomic assay into a comprehensive mutation profile along with the tumor type to be used for the analysis.
- Variant and Biomarker Interpretation: Review variants as somatic or germline and as oncogenic driver mutations interpreted as biomarkers for therapeutic, diagnostic, or prognostic evidence.
- Prepare Draft Report: Compose the patient report summary based on the interpreted variants and their clinical significance.
- Save and Sign Out: Ensure all re-usable interpretation components are saved to an internal database at the appropriate re-use scope and sign out
Step 1: Sample and Variant Quality
The number of cancer driver genes that can be interpreted as clinically actionable biomarkers is actually quite small. For most tumors, there may be only a couple dozen genes that commonly recur with Tier I and Tier II clinical evidence by the standards of AMP classification. Given the need to sequence at very high read depths (sensitivity) to detect somatic mutations from a tumor biopsy that will contain some portion on “non-tumor” cells as well as different sub-clonal groups of various stages of the tumor evolution, many cancer gene panels on the market target 50-100 genes. Newer panels shoot for the 300-500 range largely to get a better global molecular profile for metrics such as Tumor Mutation Burden (TMB).
When reviewing the quality of a sample produced by these types of panels, various summary and coverage statistics can be used to determine if the sample should be interpreted or be failed and potentially re-processed or even re-biopsied.
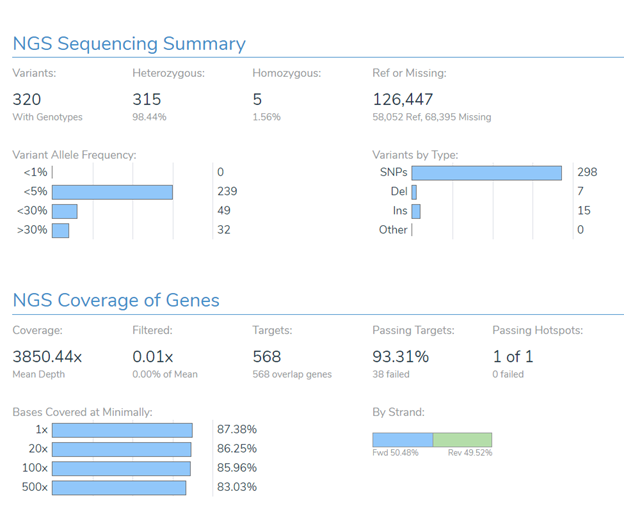
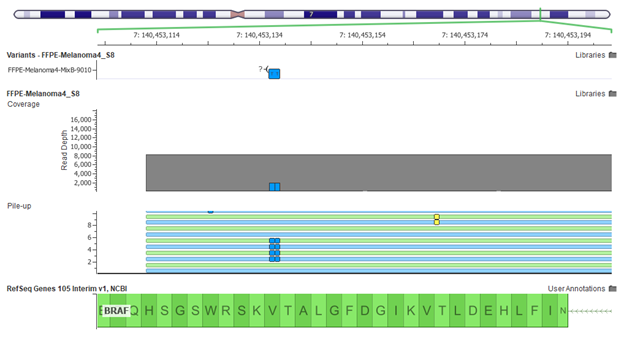
Beyond hitting global metrics such as the average read depth over the target regions, specific regions containing common mutations for a given tumor type must be reviewed to have sufficient coverage to avoid the potential for a false-negative (missed call). For example, a sample with a tumor type of Melanoma should be failed if the BRAF V600 amino acid is not sufficiently covered to allow the detection of the common and clinically actionable V600E mutation. At the same time, any exons that drop below a target threshold may need to be reviewed by hand or selected to be reported as regions of potential missed findings.
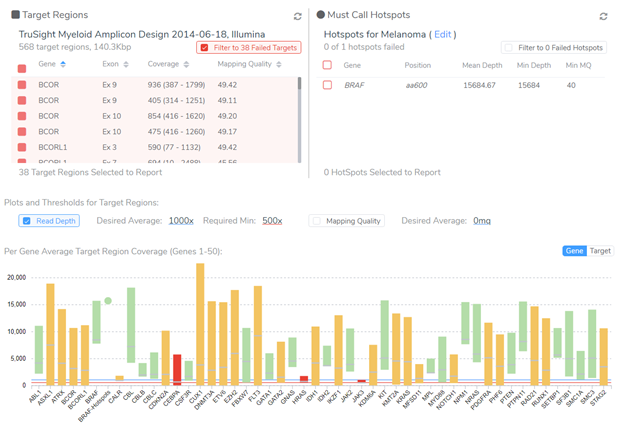
After approving the sample for analysis, the next step is reviewing the filtered small variants and CNVs in the project. VarSeq is a powerful variant annotation and filtering engine. I won’t get into the details of how to construct a somatic variant filtering strategy here, but I will note that this step is very lab-specific. Most importantly, it should reflect the specific levels of detection (LOD) of the genetic test and be able to be locked down to be part of a validated CLIA lab process. VarSeq supports these requirements and ships with a number of useful cancer-specific annotation sources such as the latest version somatic catalogs including COSMIC, TCGA, and ICGC.

After variant filtration, it is common to inspect each candidate variant in its genomic context to ensure that it is called correctly and that there are no other nearby variants that might change the clinical interpretation context.
CNVs can be called directly off of BAM files with the addition of VS-CNV; thorough CNV analysis — annotation, filtering, and review — is needed before selecting the ones likely to be clinical biomarkers.
Now that variants and CNVs are checked for quality and ready for analysis, the next step is to bring them into VSClinical and build out the patient’s molecular profile which we have covered in “Following the AMP Guidelines with VSClinical: Part II”.



