The support team at Golden Helix is always on-hand to help with your SVS and VarSeq needs. We get some questions more often than others, and this blog will answer some of the most common questions we’ve been seeing lately regarding VarSeq.
A common question we receive is if data can be filtered from a locally kept set of variants or genomic features in a BED file. The answer is – yes you can! The file doesn’t even need to be converted through the Data Source Library Convert Wizard. To set up a BED file, the minimum information needed in the file is the chromosome, and start and stop position information; the example below also includes a description of that particular genomic feature, Figure 1.
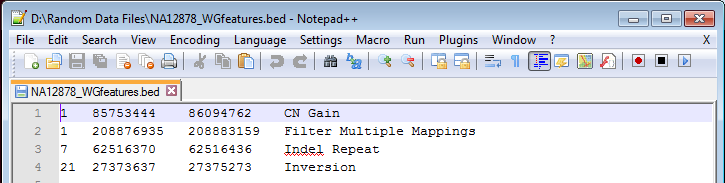
Figure 1. Example of a BED file.
Tip: When you are adding additional features to your BED file, make sure they are added in genomic order, otherwise creating the genomic index will be problematic.
The only requirement before annotating and filtering is that the file has an associated index (BED.BAI) file in the same directory. If you do not have an index for the file, you can use the Data Source Library to create it. From your VarSeq project, go to Annotate with Data Source from the variant table and select Browse to navigate to the Folder with your BED file. Then, inside the Data Source Library right click on the BED file and choose Computations on Source and select the option for creating a Genomic Index. Once the BAI file is available, you can select that file and annotate your variant table. Once the data has been annotated with your variants, you can create a filter card for the Match? column or in this case the feature of interest is listed in a second column which can also be used as a filter, Figure 2.

Figure 2. The annotated list in the VarSeq Variant Table.
Another common question we get asked is whether you can filter a dataset from a precompiled list of genes, and yes, VarSeq is capable of this too! All it takes is going to Compute Additional Data under the variant table and selecting Match Gene List. Then you need to select the gene names to match, in this case I used the RefSeq Genes 105 Gene Track. A final window then appears where you can type in or copy and paste a list of genes of interest, Figure 3.
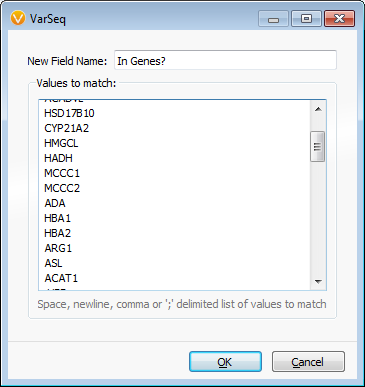
Figure 3: Gene list pasted in from external document.
The program will identify the variants found in the genes indicated in the variant table. Then you can use the In Genes? column as a filter to only see variants found in your list of genes or to selectively exclude those variants, Figure 4.
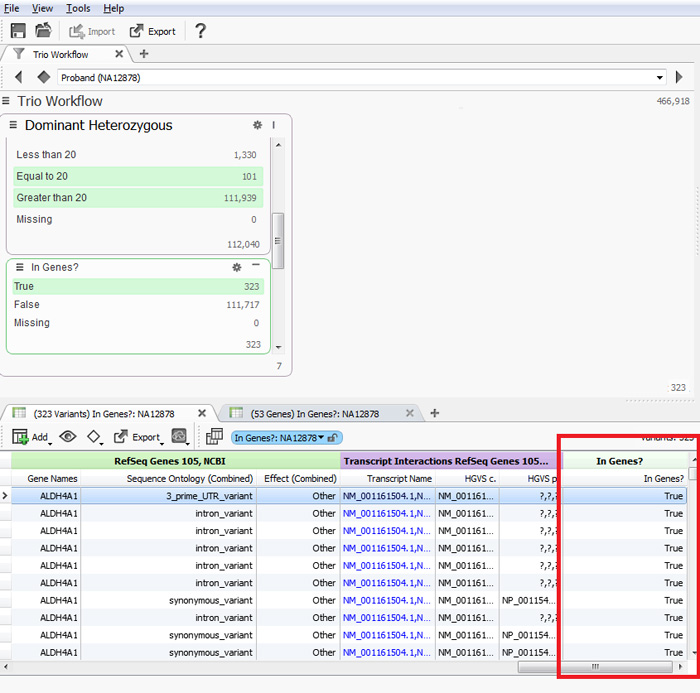
Figure 4. Variant Table with In Genes? column and example filter chain.
The final question that we know is on a lot of our customers’ minds is – when and how often are the SVS and VarSeq Annotation Sources updated? I’m happy to address that here. Sources provided in our Public Annotation repository are updated based on customer requests, as well as the release cycle of the specific data sources. For high priority data sources (ex. ClinVar, dbSNP, ExAC), we monitor the source repositories for new data releases and add them to the list of available source in SVS as soon as we are able. If you’re interested in MedGenome’s OncoMD, our newest annotation source is available as an add-on feature to your VarSeq license. You can find more information on this new addition here.
Hopefully these frequently asked questions satisfy your curiosity, but if you have additional questions about VarSeq, the Support Team at Golden Helix is happy to answer them! Email us at [email protected].



