
We’re excited to announce the release of gnomAD 4.1 variant frequencies as an annotation track in VarSeq. This latest release addresses key issues from previous versions and introduces joint variant frequencies curated directly from gnomAD. Additionally, we’ve refined our liftover process, offering more accurate GRCh37 tracks. By incorporating this data into the VarSeq data source library, we provide users with access to the most comprehensive allele frequency data available.
Updated Joint Allele Frequencies
In previous releases, we constructed our own joint gnomAD database by merging exome and genome frequency tracks into a unified annotation source. With the gnomAD 4.1 release, this process has been simplified, as the Broad Institute has provided their own harmonized joint allele frequency data. This track reports combined allele frequencies, integrating data across 734,947 exomes and 76,215 genomes. This track also introduces a new flag that indicates when a variant has highly discordant frequencies between the exome and genome databases. The figure below shows several intronic variants in the IVD gene.
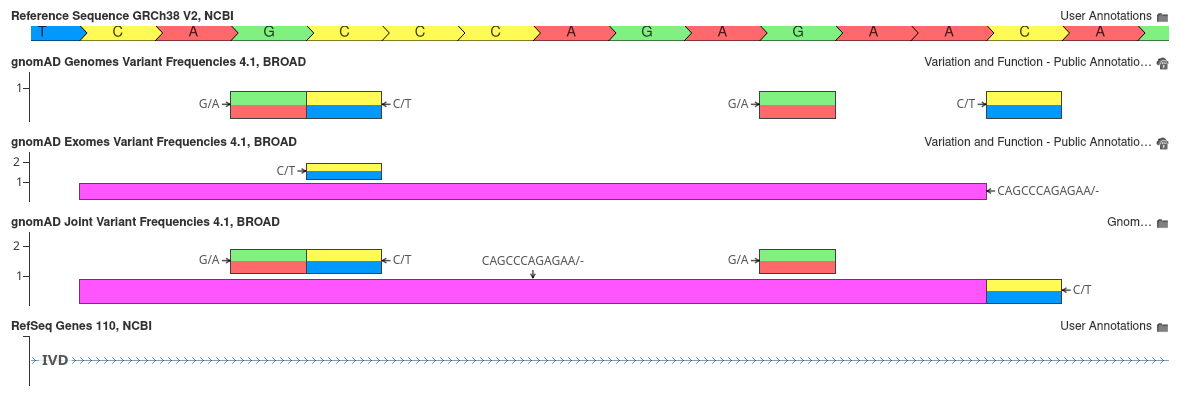
As shown, the joint track not only includes variants unique to the gnomAD exome and genome tracks, but also merges variants that are common to both tracks into a single entry. This merging combines allele counts and frequency calculations, offering a more unified view of the variant frequency data.
Improvements to the GRCh37 gnomAD tracks
Since the gnomAD 4.1 data is native to the GRCh38 reference genome, variants must be converted to the GRCh37 reference assembly using a liftover procedure. This can be particularly challenging when the reference allele differs between assemblies. While simple substitutions are straightforward, deletions often pose more complex challenges. The figure below shows an example of such a case.
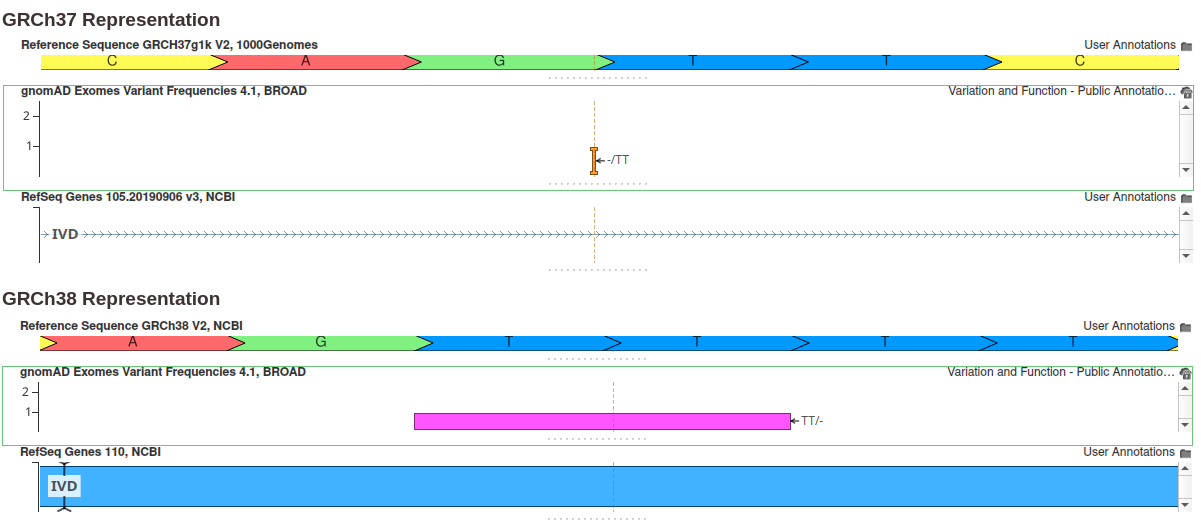
In this example, the GRCh38 reference sequence contains a repeated sequence of four Ts, and the variant is represented as a two-base deletion. However, in the GRCh37 assembly, this repeated sequence is shortened to just two Ts, and the variant is represented as a two-base insertion. Thus, the reference and alternate alleles are swapped when transitioning from GRCh38 to GRCh37, and the corresponding allele frequency values must be adjusted.
To address these edge cases, we have improved our liftover process for deletions. Specifically, when a deletion’s liftover results in a reference sequence mismatch, we cross-reference the GRCh37 Ref/Alt values from dbSNP using the variant identifier. If a single unique matching variant is found, we use the variant definition specified by dbSNP; otherwise, we add the LIFTOVER_FAILED flag to the Filter field. Although no liftover process can match the accuracy of natively aligned data, we believe the Golden Helix gnomAD 4.1 GRCh37 track offers the most precise representation of gnomAD data on the GRCh37 reference assembly.
Conclusion
We are thrilled to make the gnomAD 4.1 annotation tracks available to our customers for use in their VarSeq workflows. Both the Broad Institute and our Golden Helix curation team have worked hard to deliver the most reliable gnomAD annotation to date. If you have any questions about these gnomAD resources or how to add gnomAD to your VarSeq projects, please don’t hesitate to contact us at [email protected].



