The current reduced cost and increase availability of genome sequencing has been making academics, clinicians and individuals alike excited with the possibility of increased research depth, diagnosing capability and personal curiosity. And although a freshly sequenced genome is chock-full of tasty letter snippets, the real revelation and education occurs when comparing to an annotation foundation.
In this post, I’ll review the annotations Golden Helix provides as part of genomic analysis platforms VarSeq, SNP & Variation Suite and GenomeBrowse. We spend a lot of time digging through public data repositories to bring the best data to your fingertips, and often improving on the raw sources to make them most applicable to your analysis.
The Great Project of Genomic Science
In some ways, almost all genomic science is contributing in a small or large way back to our collective understanding of the genomes of species, and that understanding is often represented as genomic annotations.
Annotations allow one to delineate and interpret different genome regions, without which genomic data is as useful as a random number drawn from a hat. Notable annotations are accumulations of years of researcher’s work, the product of large collaborative consortiums and the dedicated efforts on specialized curators such as those at the National Center for Biotechnology Information. Different sources emphasize different genome features from the chromosomal level down to that of a single nucleotide polymorphism (SNP).


Each of these annotation sources can consistently improve the analysis experience by increasing the richness of the understanding of a genomic variant, the local genomic region or the referenceable clinical and phenotype context.
Curate, Wrangle, Validate, Repeat
Golden Helix invests a lot of resources into curating both a comprehensive and high-quality catalog of annotations.
The curation process starts from acquiring the latest raw data from the annotation source. From there, it translates it to a form that works easily within the Golden Helix suite of analysis software products and finally it makes the nuances of each annotation source more easily recognized and applied. Finally, each source is validated in its final form both against the other similar sources and against previous versions to detect anomalies in the deltas.
A list of notable annotation sources that are annotated and curated by Golden Helix are listed below.
- Gene Sources:
- GRCh37: RefSeq Genes 105 (Updated with the “Iterim” release from March, 2017)
- GRCh37: Ensembl Genes 75
- GRCh37: GENCODE Genes 19
- GRCh38: RefSeq Genes 108
- GRCh38: Sensembl Genes 84
- Gene Based:
- OMIM Gene annotations including linked phenotypes and their inheritance model
- ClinGen Dosage Sensitivy
- COSMIC Cancer Genes
- ExAC Function Gene Constraints (which gene tolerate mutations)
- DbNSFP aggregate annotations on genes
- Variant Population Catalogs:
- gnomAD Exomes (123K samples) and Genomes (15K samples)
- ExAC Exomes (61K samples)
- 1000 Genomes (2.5K samples)
- NCBI dbSNP
- UK10K Twins and ALSPAC Populations
- Supercentenarian 17 Variant Frequencies
- Clinical Interpretation:
- NCBI ClinVar
- OMIM Variant and Genes
- GWAS Catalog
- ClinVitae Clinical Assertions
- Cancer Specific Annotations:
- MedGenome OncoMD Variation and Gene Summaries
- MedGenome OncoMD Drug Targeting Mutations
- CIViC Clinical Evidence for Somatic Variation and Targeted Molecular Therapy
- ICGC Simple Somatic Mutation Catalog (21 cancer sites)
- COSMIC Mutation Catalog
- Variant Function:
- dbNSFP Functional Predictions and Conservation Scores
- dbscSNV Splice Altering Predictions
- CNV Annotations:
- ExAC XHMM CNV Calls
- Database of Genomic Variants (DGV)
- NCBI ClinVar CNVs
- 1000 Genomes CNV Calls
While all data sources start from a bulk download from their original source such as NCBI or the Exome Aggregation Consortium (ExAC), from there each source is treated uniquely. Some sources are on a scheduled release cycle. Our monthly updated sources include OMIM, OncoMD, ClinVar and CiVIC. Other sources are updated based on the release schedule of the research consortium or group that publishes them.
The raw data is parsed and transformed through various normalization and clean-up steps with a custom code using SVS, and then passed into VarSeq for labeling and documentation.
VarSeq is used as a variant analysis tool, but is also a Swiss army knife for variant set manipulation, comparison, clean-up and documentation. Per-field documentation and descriptions of each category, categorical fields are carefully added. This information enriches the analysis process, and shows up in VSReports and other downstream parts of the clinical interpretation experience.
The final step is to compare the current annotation to the previous versions or similar annotation sources to verify the changes are successive and do meet expectations.
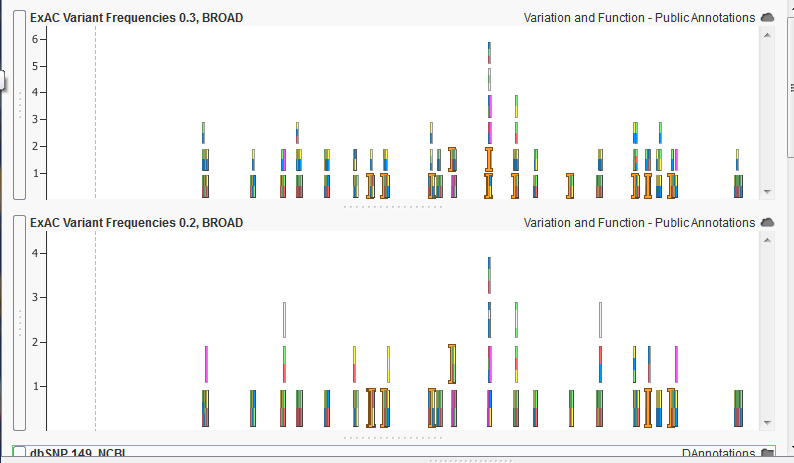
The example above shows a comparison between two versions of the ExAC annotation track. While both show very similar behavior, the more recent version on the top shows more color blocks which represent more known allele frequency at the given location.
This variety of valid annotation sources with variable application requires reliable cataloging and updating. We are continuing to add new annotation tracks as well as support for new species. If you would like to request a specific database to be converted into an annotation track, or would like to see a particular track in SVS, VarSeq or GenomeBrowse, please email us and let us know.



