
Discover how soft clip visualization can help you identify structural variants in your research and improve the accuracy of your findings.
Soft clipping is a common technique in sequence alignment used to remove bases from the ends of reads that do not align with the reference sequence. Removing these bases typically improves alignment accuracy. However, when multiple reads are soft clipped at the same location, it can indicate a structural variation in the sample. Visualizing these regions is crucial when performing quality control on structural variant calls.
Coverage
With the latest release of Genome Browse and VarSeq, soft clipped sections of reads can now be visualized when plotting BAMs and CRAMs. To enable this, uncheck the “Hide Soft Clipped Bases” option in the plot controls.:
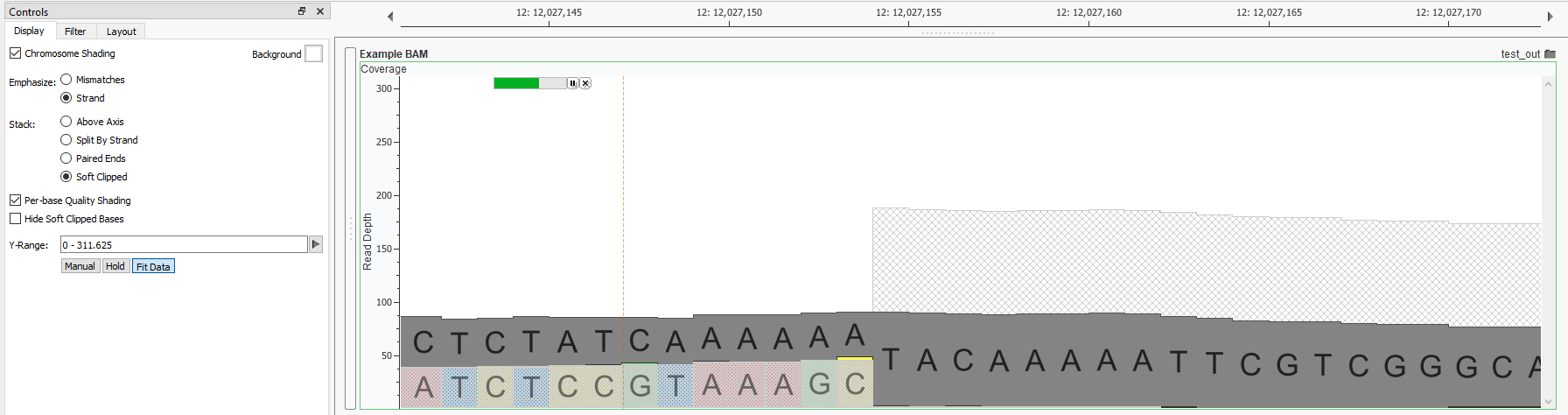
In the example, a distinct cliff of well-aligned reads is visible on the right, while half of those reads on the left become soft clipped and are displayed with a faded base color. By plotting the base and color for the soft clipped read, it becomes evident that most of the soft clipped reads are at the same base in this region, indicating a strong likelihood of a structural variant. In this case, the soft clipped reads bridge the fusion of the right-handed segment to another region on the genome. Clicking the first base in the soft clipped region (C) displays the base coverage statistics in the console view for this break point.
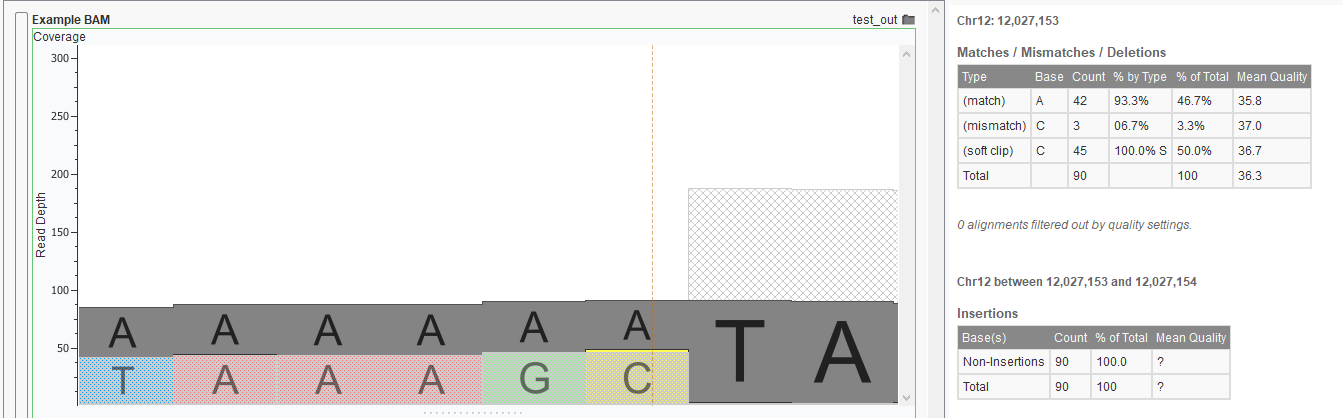
In this example you can see the mean quality, as well as the depth for the different base calls at this location. In this example, there are some reads which have a mismatched C at this location; perhaps they should have been a soft-clipped instead.
Pileup
Also new is the ability to collate the reads by soft clipped status. This makes it easy to compare those reads which were soft-clipped and spot check events called at these locations. If their mate falls on the soft-clipped side and is in the same location these reads are likely part of the same event.
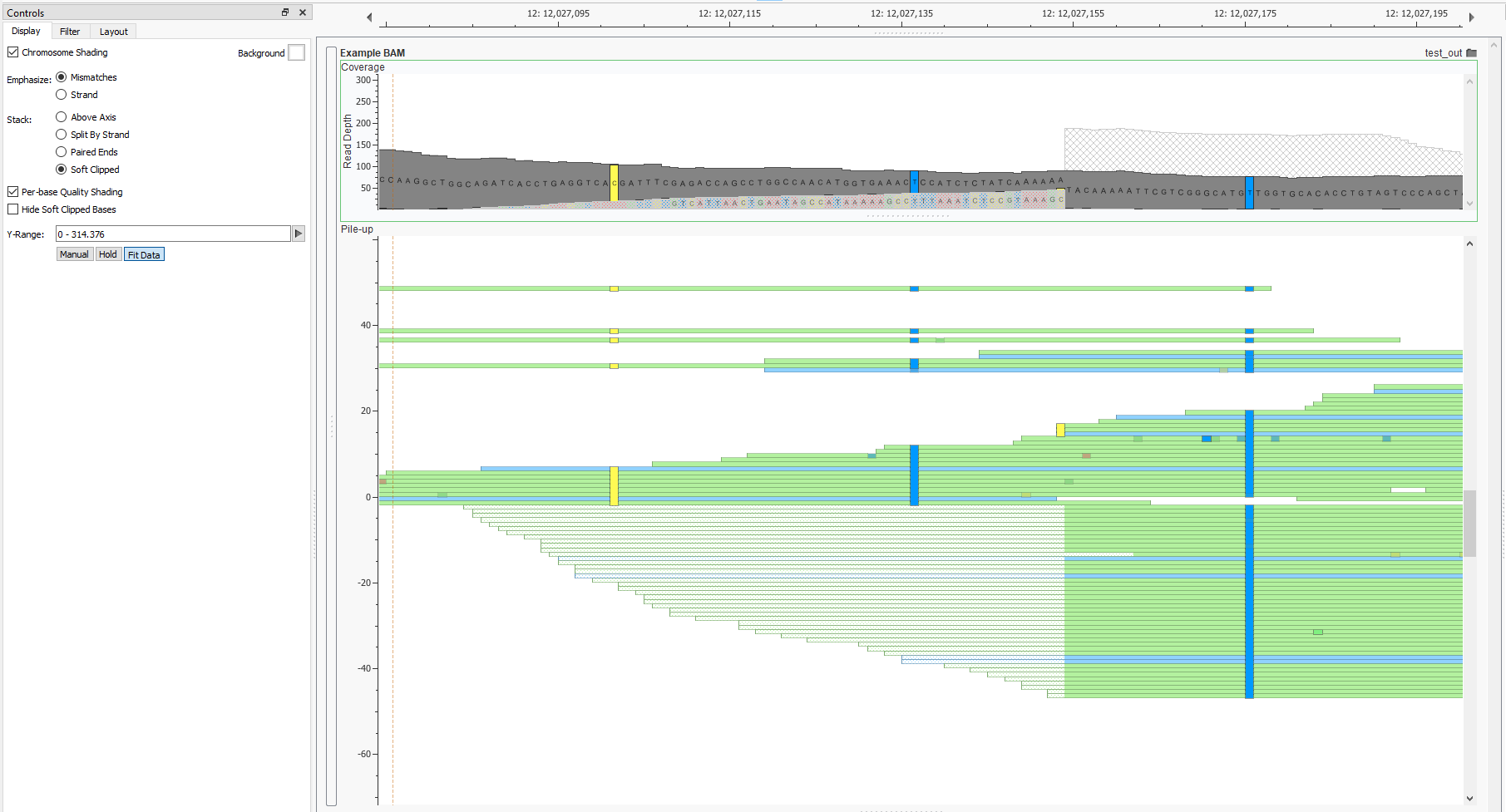
This option can be found in the “Stack” section of the BAM/CRAM plot controls. This separates the reads placing those with soft clipped tails bellow the the center line and those without above. This is a helpful way to see the distribution of the clip point in the reads. In this particular case you can see the position of the clip point is well distributed on both the forward and reverse strand. This further supports a structural variant call at this location.
With the latest release of Genome Browse and VarSeq, visualizing soft clipped sections of reads is now easier than ever, allowing for more effective quality control of structural variant calls. If you need further assistance or have any questions about these new features, please don’t hesitate to contact support at [email protected].



