Last month, June 2014, we announced a new method that Golden Helix developed–the soon to be available MM-KBAC, a statistical advancement built into our genomic analysis software. MM-KBAC, or Mixed Model Kernel Based Adaptive Clustering combines the KBAC method developed by Lui and Leal (2010) with a random effects matrix to adjust for relationships between samples. The KBAC algorithm takes a binary dependent variable and transformations are used to convert the logistic regression model to a linear model so that EMMAX (Efficient Mixed Model Association eXpedited) can be used to solve the equations.
We are also very excited that we have been accepted to present this material at ASHG this October (we’ll be in booth 422)! More importantly, we will be making this method available to our customers with the next release of SVS due out in August.
What do you need to know about this method?
- This method is designed to correct for batch effects, cryptic relatedness and known structure from large pedigrees in large-N DNA Sequencing studies.
- The KBAC algorithm identifies genes with an increased burden of rare variants associated with the phenotype, complementing standard variant analysis pipelines.
- You can use this method with data from plants, animals or humans; as long as your data comes from a DNA Sequencer (or an Exome chip designed for rare variants) and you have a gene annotation source for your species. This makes it equally applicable to exome analysis workflows targeting rare coding variants.
- The p-values are determined via permutation testing so the number of permutations performed will determine the smallest p-value possible for the dataset.
How will you use MM-KBAC?
Like with EMMAX or Mixed Linear Model Analysis, you first need to have a kinship matrix for your samples. Kinship matrices are more accurate if only COMMON variants are used to determine the allelic sharing between samples. Once you have filtered your variants to exclude any rare variants, you can compute either an IBS (Identity by State) matrix or GRM (genomic relationship matrix) to use as the kinship matrix. Note: it is much faster to compute a GRM and not much is lost by doing so. In the next release, you will also have the option to explicitly specify the kinship matrix based on pedigree information if you have that available for all of your samples.
After computing the kinship matrix, the original variant list will need to be filtered again, this time leaving only the rare variants. Now, you are ready for MM-KBAC. With a quick selection of the gene annotation source, the parameters for KBAC, the kinship matrix and any other covariates, you are off to correcting your large-N DNA Sequencing data for pedigree structure to find those genes with a burden of rare variants associated with the phenotype. MM-KBAC is particularly valuable in NGS data analysis workflows where population structure and relatedness can confound rare-variant burden tests.
With one click of the button, you can obtain all the results you will need!
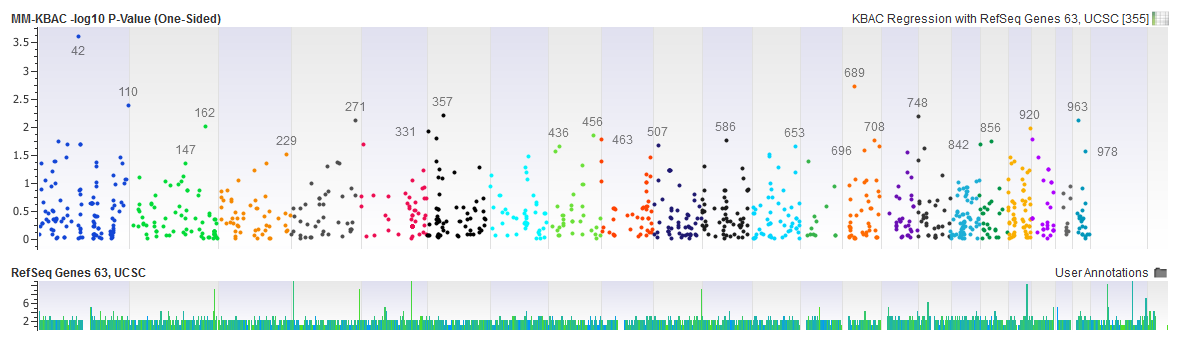
Let us know if you would like to try this out for yourself and cannot wait until the August release; we would be happy to provide you with the beta release. You just need to be an active SVS customer and willing to give us feedback on how this new feature functions with your data.




Dear Greta Linse Peterson,
I attended your webcast which was about MMKBAC several weeks ago. I have few questions about this method as well as SVS. It seems that MMKBAC only takes binary trait (is that right?), because you use a logistic mixed model. I’m wondering that do you have any similar methods (mixed model + gene-based test) implemented in SVS which can take continuous traits?
Thanks,
Meijian Guan
CMC with Regression is our gene-based collapsing test that can take a continuous trait. We have plans to add in mixed models to CMC with Regression as well as add in the logistic mixed model to EMMAX.
If you would like more information on what we plan to implement in the future as well as how to analyze your continuous trait with your data please email [email protected].
Best,
Greta