As VarSeq is quickly becoming the go-to variant analysis software for tertiary analysis, we want to give our readers the opportunity to examine completed projects from start to finish. As an addition to the currently available demonstration projects, we are pleased to provide users with a Single Exome Analysis example project.
To access this project simply click here to download the VarSeq example project. Not an existing VarSeq user but interested in viewing all the annotations, algorithms, and filtering structure applied to this workflow? You can download VarSeq Viewer! Once the viewer is downloaded you can view the Single exome project along with three other projects visible in the welcome window (Cardio gene panel, Tumor/Normal analysis, Trio analysis).
The Single Exome Analysis example project examines a single sample (NA19240) from the Yoruban population from the HapMap Project. BAM files came from the 1000Genomes Phase3 Illumina Exome Alignment Project. We injected a variant into the daughter’s BAM file and then used Sentieon’s powerful secondary analysis tools for variant calling. The injected mutation is in the SMAD4 gene, which is associated with Myrhe Syndrome. Upon importing the data, We’ve imported variants annotated as “Pass” by the variant caller and limited potential off targets to +/- 5 bp outside the exon boundary.
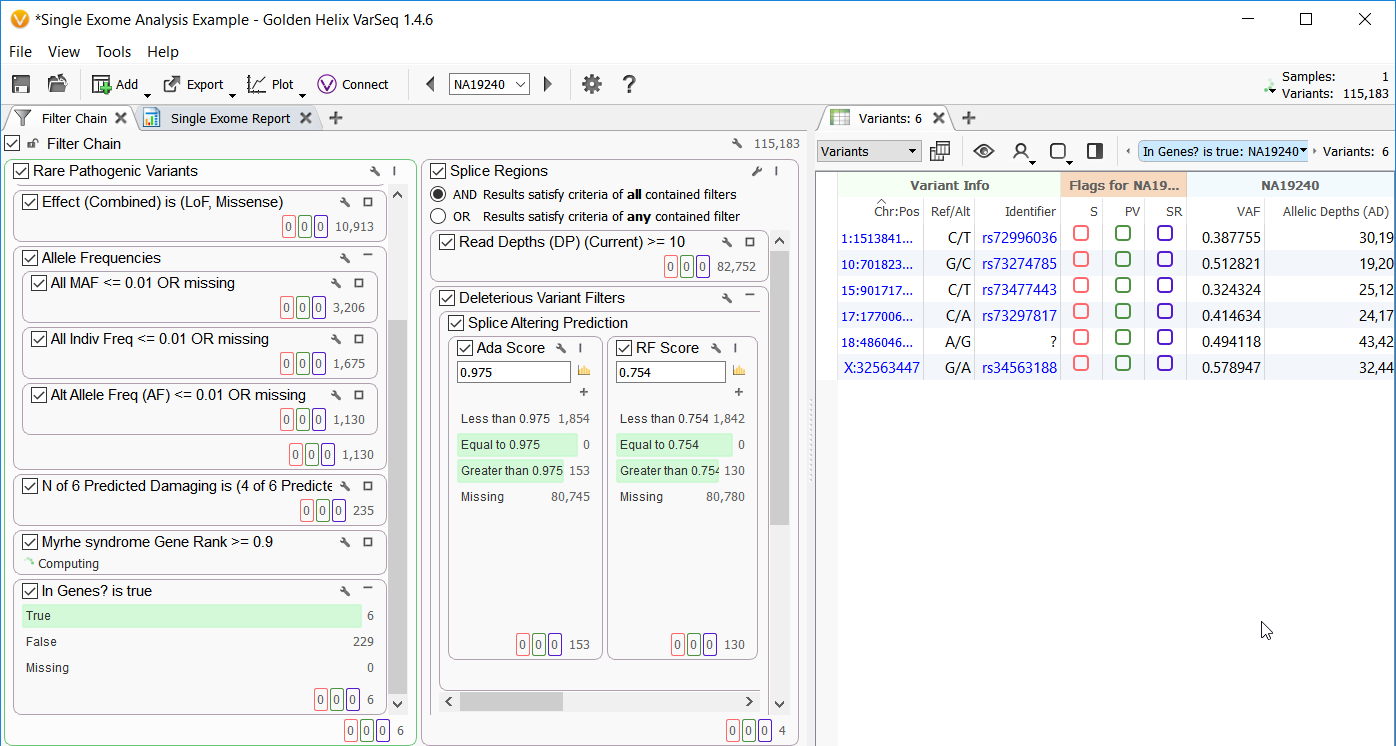
Figure 1. Project view for Example Project Single Exome. (Click image to enlarge).
Once you have opened the project you will notice a fully annotated variant table and two elaborate filter chains which analyze rare pathogenic variants and potentially damaging splice site variants (Figure 1). This is accomplished by changing the logic operator inside the filter view, switching it from “AND” to “OR” and then adding multiple filter containers to build each separate workflow. The union of each workflow is then seen at the bottom of the filter view for further analysis.
Looking for Rare, Non-Synonymous Variants
In this project, there are 6 unique variants identified by the Rare Pathogenic Workflow. This workflow aims to:
- Remove any low-quality variants by filtering on Read Depth and Genotype Quality scores;
- Removing common variants by filtering on allele frequency in population catalogs (1 kG genomes, NHLBI, GnomAD);
- Identify any mutations/variants with a predicted missense or loss of function effect according to our transcript annotation algorithm; and
- Examine variants in genes that match my gene list and are associated with phenotypes that are characteristic of Myhre Syndrome (intellectual disability and global developmental delay).
As a reminder, every column in VarSeq can be utilized as a filtering criteria simply by right-clicking on the column header and selecting “Create a Filter” (Figure 2). With so many annotations available at your fingertips, VarSeq enables users to create simple and intuitive workflows allowing users to deep-dive into each exome sample.
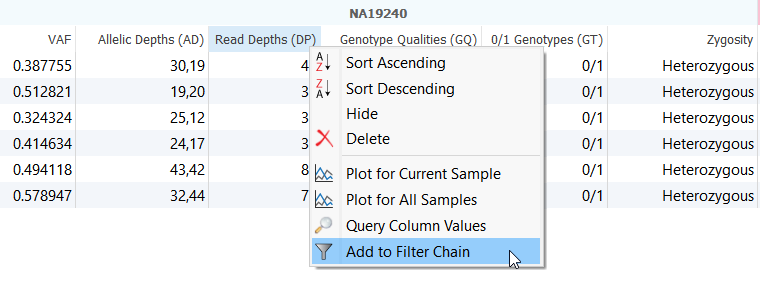
Figure 2. Creating a Filter Card.
With this in mind, let’s take a closer look how this filter chain was constructed.
The first two filter cards are filtering out low quality variants using the sample level fields imported from the VCF file (Figure 2). Next, you may want to start by computing the Zygosity State of the variants in the data. The zygosity state feature is helpful if you are aware of the type of variant you are looking for. Homozygous would be relevant for a recessive phenotype and removing reference alleles is beneficial if multiple samples were called together in secondary analysis.
Following the removal of low-quality variants and reference calls, next we are going to look for non-synonymous variants. In this case, we know the variant has a severe phenotype so using a gene track such as RefSeq to gain sequence ontology information would be beneficial. The sequence ontology information can be used directly or when annotating against a gene track. Taking a closer look at the fields within we can see the relevant sequence ontology and the “Effect” of each sequence ontology term. The “Effect” column bins each sequence ontology into three categories for ease of filtering (Figure 3):
- Those variants that will likely cause the transcripts product to lose function are categorized as “Loss of Function” (LoF);
- Variants which will cause at least one amino acid change are categorized as “Missense”; or
- Finally those variants which likely have little on no effect on the functional transcript as categorized as “Other”.
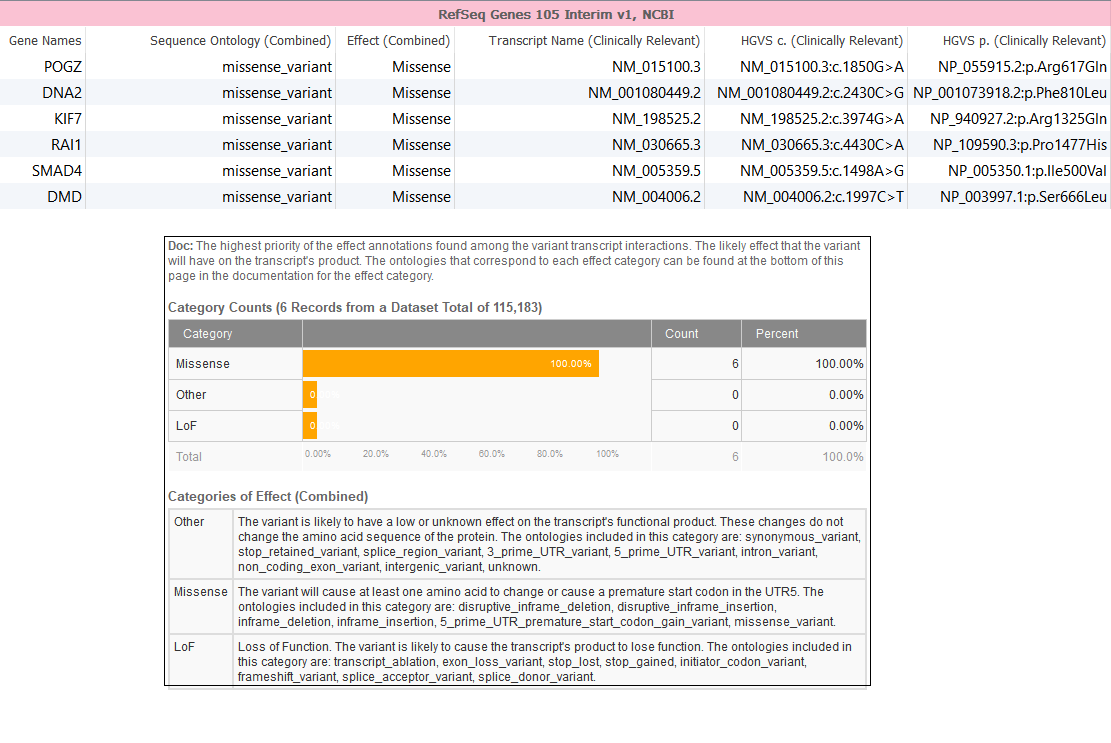
Figure 3. Annotating variants with RefSeq gene(s).
In this case, the Effect simplification was used and LoF and Missense variants were kept.
This particular individual presented clinically with global developmental delay and facial abnormalities. Based on the family information, it seems to be caused by a rare variant. Therefore common variants found in the population can be removed. Using population level allele frequency data from NHLBI, 1k Genomes, and gnomAD databases, variants higher than a 1% frequency were removed (Figure 4).
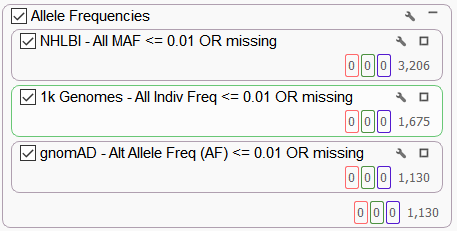
Figure 4. Filtering for Rare Variants According to NHLBI, 1k Genomes, and gnomAD Variant Frequency Tracks.
The last filter derived from our annotations was from dbNSFP 3.0, a functional prediction database. This track includes the pre-computed functional predictions for five different algorithms identifying if variants are damaging or tolerated: SIFT, Polyphen2, MutationTaster, MutationAssesor and FATHMM. VarSeq tabulates these sources into the number of algorithms that consider a variant damaging or tolerated, so you can select how many out of those five consider it damaging. Also, each of the different algorithms’ results can be used for filtering if you have a favorite algorithm in mind (Figure 5)!
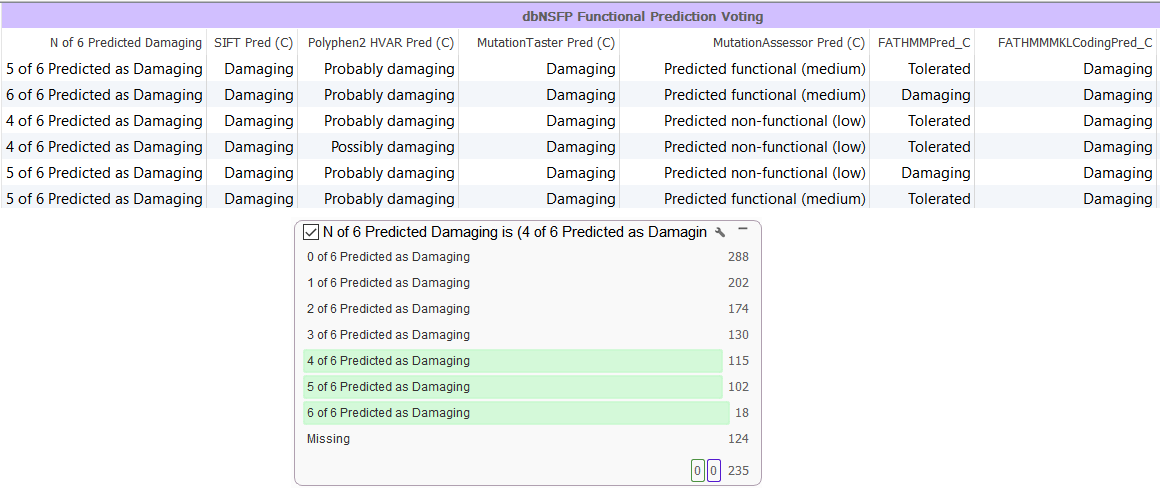
Figure 5. Utilizing Functional Prediction Algorithms in VarSeq.
Rank Genotypes and Match Gene Lists
The final two filters in this workflow are derived from two of our popular algorithms; Phorank and Match Gene List.
The PhoRank algorithm works by leveraging the knowledge stored in biomedical ontologies, such as the Human Phenotype Ontology (HPO) and the Gene Ontology (GO). These ontologies model relationships between genes and diseases as directed acyclic graphs which ultimately gives you the ability to associate phenotype terms from the Human Phenotype Ontology to the genes in your dataset. It may be useful to see how your genomic data is linked to specific phenotype terms at various points of the analysis. In this case, we are applying PhoRank after using a variant annotation algorithm to filter variants down to certain functional and population frequency criteria.
In this project, we wanted to look for variants with relevance to the following HPO terms:
- HP:0001249: intellectual disability; and
- HP:0001263: global developmental delay
Using these two search terms, the results show a Gene Rank, Gene Score and Path, described below (Figure 6).
- Gene Rank: Percentile rank of the specific gene.
- GeneScore: The score of the gene computed by the ontology propagation algorithm.
- Path: A shortest path from the gene to one of the specified phenotypes (there may be many paths to the phenotypes).
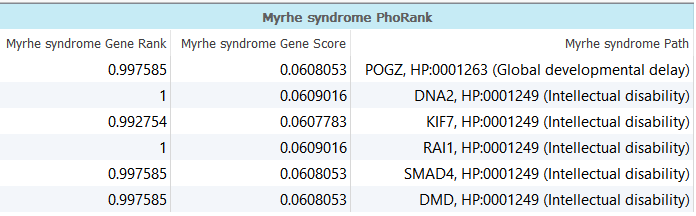
Figure 6. Leveraging the Human Phenotype Ontology (HPO) and Gene Ontology (GO) databases using PhoRank.
By filtering on the “Myhre Syndrome Gene Rank” column and selecting a high threshold you can get a quick sense of which genes possess the closest association with the imputed HPO terms.
Lastly we can further reduce our list of variants by using the Match Gene List algorithm, to determine if the variants in the data are contained within our short list of genes entered. The result of the algorithm will be a column that can then be used to filter your data to only those variants of interest (Figure 7).
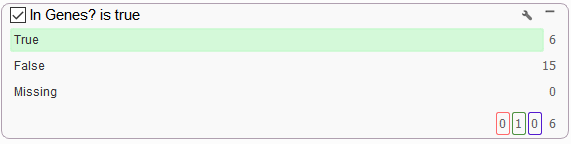
Figure 7. Looking for Variants that Match our List.
Clinical Reporting in Just a Few Clicks
These filter criteria managed to quickly sift through ~115k variants to output 6 variants of interest. This patient was found to possess a pathogenic variant in the SMAD4 gene. The mutation of SMAD4 is associated with Myrhe syndrome. Contained in the variant table is information from OMIM which includes relevant case studies that describe this variant and the associated disease. All of this information could be relevant in a clinical report (Figure 8).
Clinical Reports in VarSeq are fully customizable and can include information such as: Sequence Ontology, Coverage Information, Pathogenicity determinations, as well as information from pulled in from annotation sources such as OMIM and exAC. Here you can see how VarSeq makes it easy to get to a clinical report. After entering in the desired information for each variant the report can be created and exported from VarSeq in a PDF.
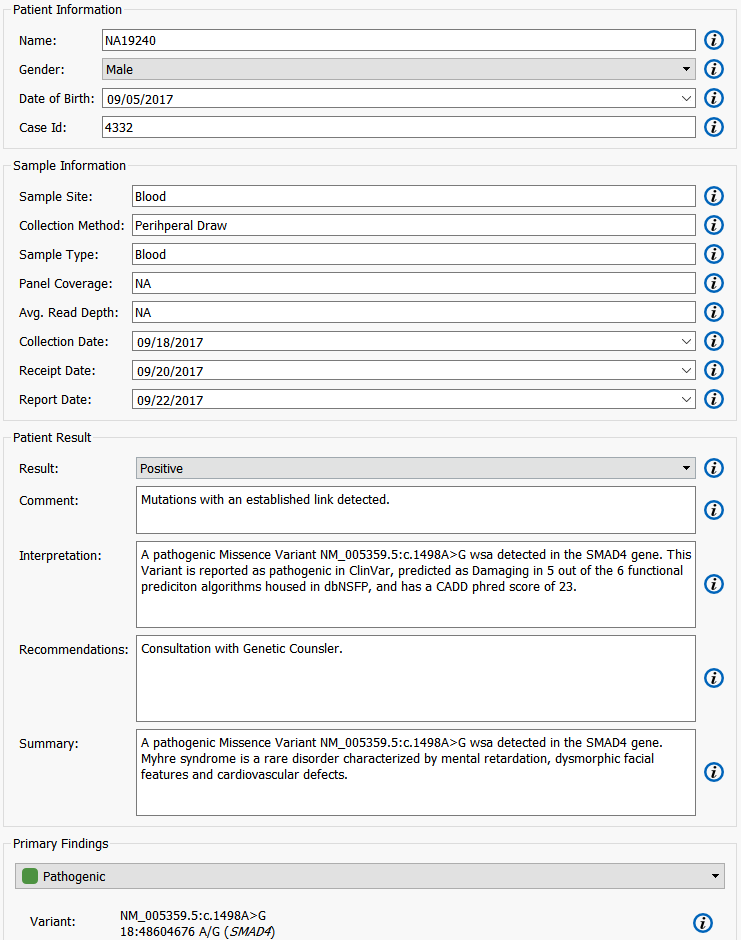
Figure 8. Creating A Clinical Report Within VarSeq.
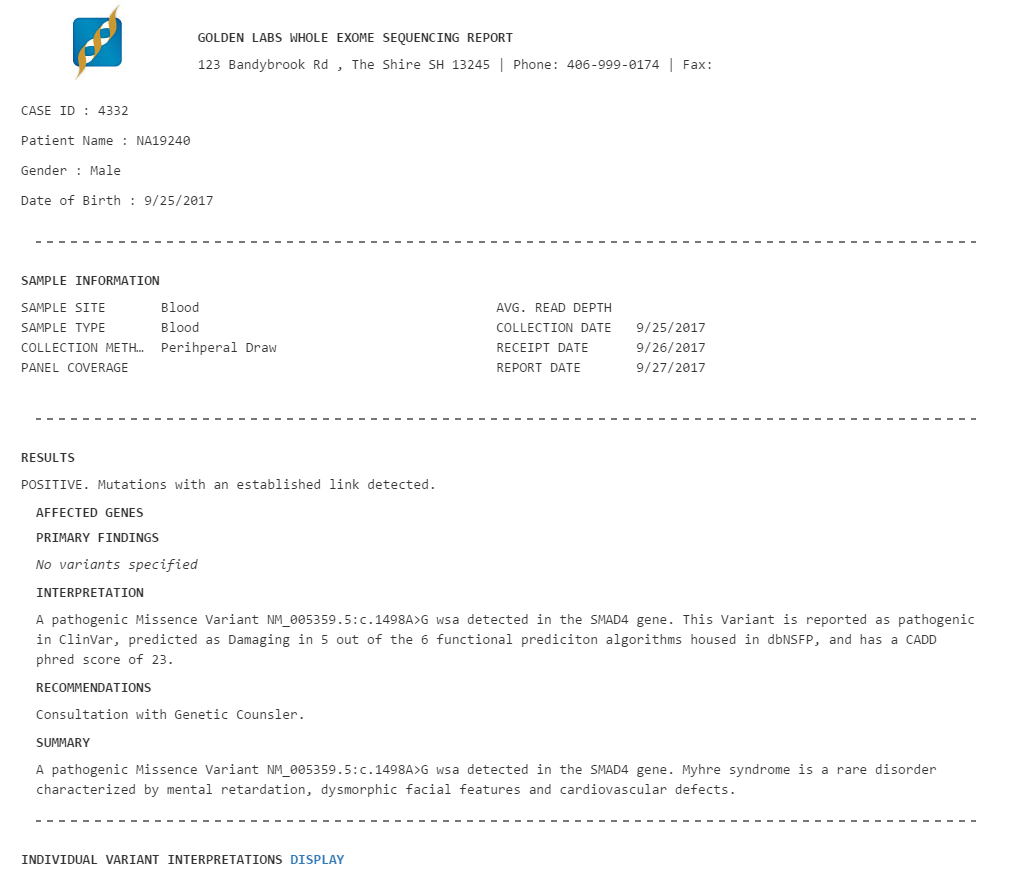
Figure 9. One Example of our Many Custom Report Templates.
Sugar, Splice, and Everything Nice
Interested in other potential workflows? There are many approaches that one might use to define a variant as potentially deleterious. In addition to workflows based on rare, non-synonymous variants, VarSeq users often ask about annotation sources to assist in the interpretation of non-coding regions. dbscSNV is a useful annotation source that may help you start to move beyond the coding exome by considering some of its nearest neighbors: splice site variants.
In addition to the “Rare Pathogenic Variants” workflow we have implemented, we have also created an excellent workflow that aims to capture data for splicing regions. This example workflow aims to leverage the ensemble scores “ADA” and “RF” and functional prediction tool CADD scores to assess potentially damaging variants occurring in the splicing consensus regions. The predicted damaging variants tend to be rare, which may be expected given the conserved nature of splice site sequences (Figure 10).
The dbscSNV annotations clearly have the potential to help VarSeq users identify potentially deleterious variants that won’t always be captured in exome filtering workflows. In this case the VarSeq filter chain is configured to select splice altering variants together with rare functional variants. As always, the user has ultimate control of how the filters are configured (Figure 10).
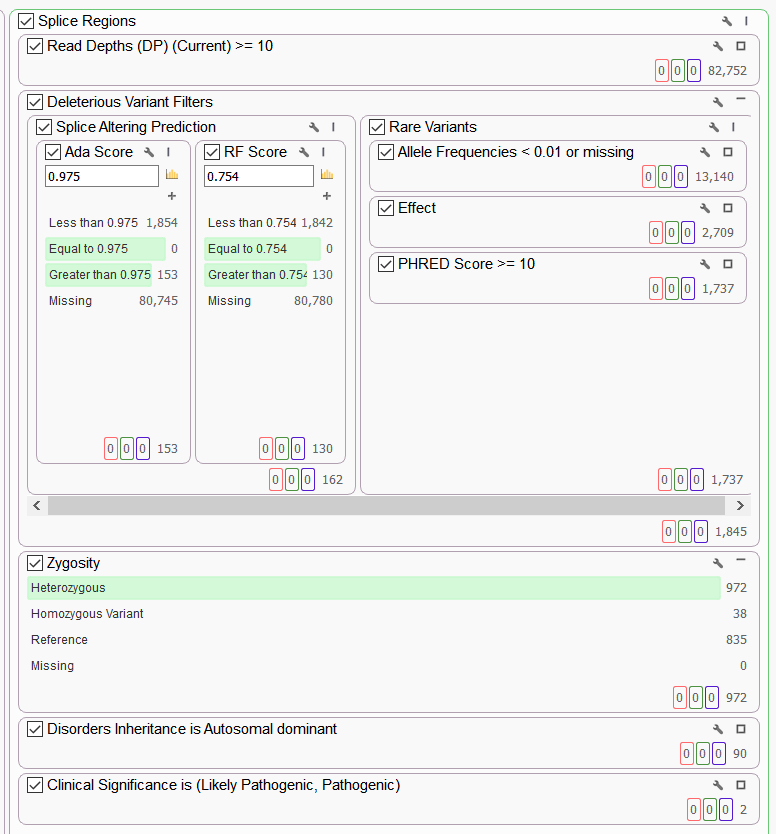
Figure 10. Analyzing Splice Site Variants in VarSeq.
VarSeq was designed to provide researchers and clinicians with an easy-to-use software that has a user friendly GUI for variant annotating and filtering. The example workflow in this blog just scratches the surface on the annotations and computations available in VarSeq. If you’re interested in trying out VarSeq for your research or clinical testing lab, please contact support@goldenhelix.com!