In this blog post, we will explore the nuances of variant calling and import in the context of VSPGx. We will discuss the importance of integrating must-call variant definitions into the calling processes and provide guidance on incorporating copy number variants (CNVs) and structural variations (SVs) into your PGx analysis workflow.
Must Call Variant Files
To ensure the optimal performance of the variant detection and recommendation capabilities provided by VSPGx, it is crucial to configure the variant calling process during the secondary analysis step to detect all variants across the full spectrum of desired pharmacogenomic (PGx) genes. Failing to capture these variants comprehensively can lead to incomplete or inaccurate diplotype determination, subsequently impacting the reliability of drug treatment recommendations.
To assist with this process, Golden Helix provides a list of must-call variants, which are available as both annotation tracks and vcf files. These files contain all variants with recommendations provided by the Clinical Pharmacogenetics Implementation Consortium (CPIC) database. The must-call variants vcf can be used as input into the variant caller to ensure that all relevant variants are called. If you are using Golden Helix for variant calling, a flag can be set to specify the must-call variants vcf as an input to the variant caller.
If you opt not to force-call these variants but are generating vcf files with “gap regions,” the VarSeq importer has a Force Call Variants option that can be selected on the final page of the import wizard.
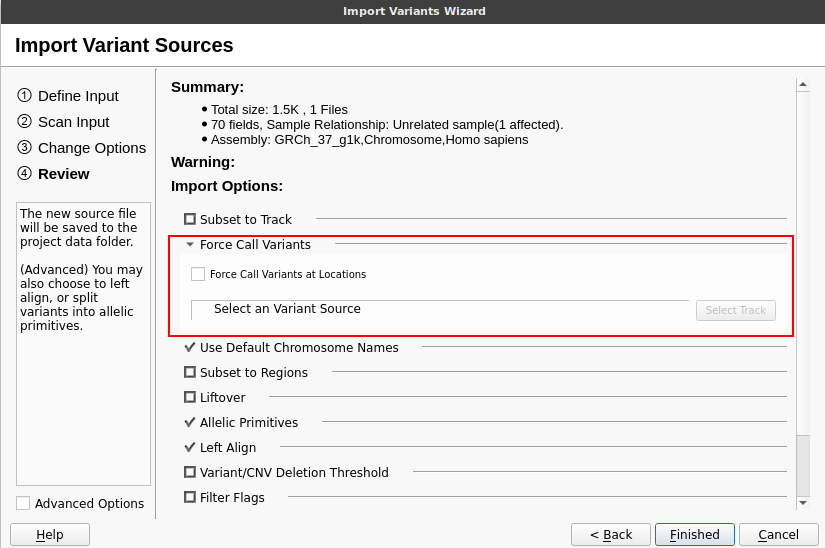
This feature allows you to select an annotation track specifying the list of must-call variants. Variants at these locations will be added to each sample on import, filling in read-depth information from the input gvcf. The must-call variant annotation track file available in our Data Repository can be used for this purpose.
Importing CNVs and SVs for PGx Analysis
CNVs and SVs commonly influence the inferred phenotype of certain genes, and robust CNV analysis is essential for accurate diplotype calling. For example, CYP2D6 phenotype prediction is sensitive to SVs and CNVs, with the CYP2D6 ultrarapid metabolizer phenotype being predicted based on duplications of normal function alleles. Additionally, both the complete deletion of CYP2D6 (*5) and the fusion of CYP2D6 with CYP2D7 (*13/*68) are considered no-function alleles.
While VSPGx does not currently identify the presence of SV and CNV alleles from vcf data, these alleles will be considered by the annotation algorithm if they are specified in the sample manifest as integer fields. To utilize this functionality, the name of each CNV field must match the name of the relevant PGx allele. For duplications, the value corresponds to the allele’s copy number, while for deletions, the value corresponds to the number of deleted copies. An example sample table with the specified CNV alleles is shown below.
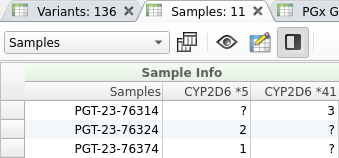
In this example:
- The first sample (PGT-23-76314) has a duplication of the CYP2D641 allele (41×3).
- The second sample (PGT-23-76324) has a homozygous deletion of the entire CYP2D6 gene (5/5).
- The third sample (PGT-23-76374) has a heterozygous deletion of CYP2D6 (1/5).
If you want to learn more about VSPGx, you can check out our webcast, where we comprehensively cover the product’s capabilities, including the ability to identify actionable pharmacogenomic diplotypes and generate clinical reports. If you are currently looking for pharmacogenomics software and would like a trial, please reach out to us at [email protected].



