In the past couple of weeks, the topic of the Filter and Quality fields in the popular ExAC population catalog has come up a number of times in discussions around NGS analysis software workflows. It turns out that unlike the 1000 Genomes project, which decided to very heavily filter their variant list to only contain variants they consider high quality, ExAC chose to include more dubious variants but to flag them with various QC checks that they failed.
This is a fine decision on their part, but it requires users of the ExAC catalog to treat any QC flagged variant with high levels of suspicion. In fact, you should consider any variant in your data that matches a QC flagged variant as if it was not in the catalog at all. (Edit: See comment below from Dan MacArthur, PI and team lead for ExAC/gnomAD, on a better phrasing of this qualification)
To make this case, I will cover a rare pathogenic variant in my own exome that is truly novel with regards to public population catalogs, but shows up in ExAC as a false-positive QC flagged variant.
The Variant That Wasn’t
With the massive new set of clinical assertions added to January’s ClinVar by Illumina, I thought it might be a good time to revisit my own exome’s variant analysis.
Using VarSeq, this is quite a straight forward process for exome analysis. I can just open up my existing personal exome analysis project and follow the notification in the top-right of the project window to update all annotations to their latest versions (including ClinVar).
With the latest annotations, a variant I possess in a heterozygous state caught my attention: NM_002769.4(PRSS1):c.86A>T (p.Asn29Ile) is listed in ClinVar as Pathogenic for Hereditary Pancreatitis in a gene that has been shown by OMIM to act in a dominant model. This means my single mutated copy would be sufficient to cause the disease!
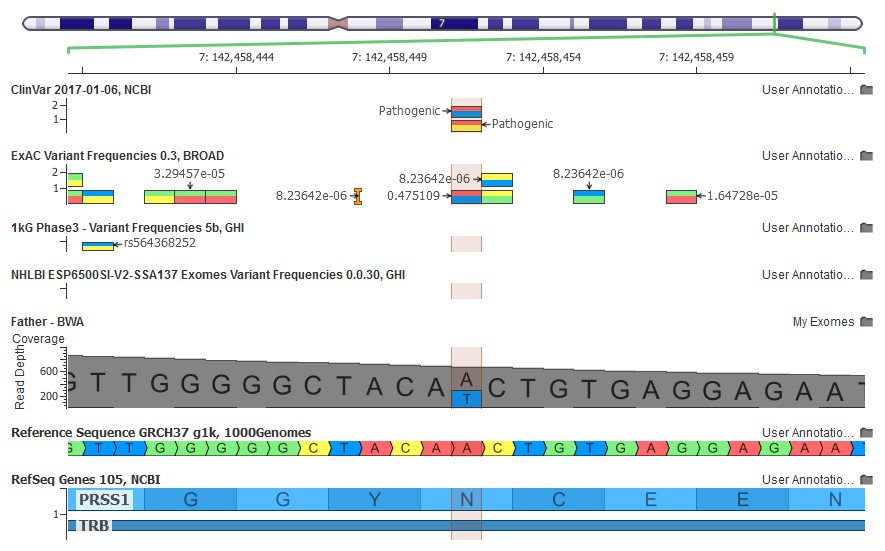
Under the Summary Evidence tab in ClinVar, Illumina provided a very thorough clinical analysis of the variant and its relationship with hereditary pancreatitis (abbreviated here):
Across a selection of available literature, the c.86A>T (p.Asn29Ile) variant, also referred to as p.Asn21Ile, has been reported in at least 160 hereditary pancreatitis (HP) patients (Gorry et al. 1997; Ferec et al. 1997; Otsuki et al. 2004; Sahin-Toth et al. 2006; Lee et al. 2011; Wang et al. 2013).
[…] In addition, in a review of a variant database maintained by Leipzig University in Germany, Sahin-Toth et al. (2006) observed that the p.Asn29Ile variant was the second most common PRSS1 variant associated with HP, accounting for approximately 25% of all identified pathogenic alleles in PRSS1.
The p.Asn29Ile variant was absent from 382 controls and is not found in the 1000 Genomes Project, the Exome Sequencing Project, or the Exome Aggregation Consortium.
[…] Based on the collective evidence, the p.Asn29Ile variant is classified as pathogenic for hereditary pancreatitis — a conclusion that underscores the importance of rigorous variant classification workflows.
If you carefully examine the GenomeBrowse screenshot above, you will notice that although Illumina claimed this variant was absent from ExAC, it actually showed up with a 47% allele frequency!
A Decoy that Improves Alignments
Zooming out of my GenomeBrowse window made me start to suspect the validity of alignments and variant calls for my exome in this gene.
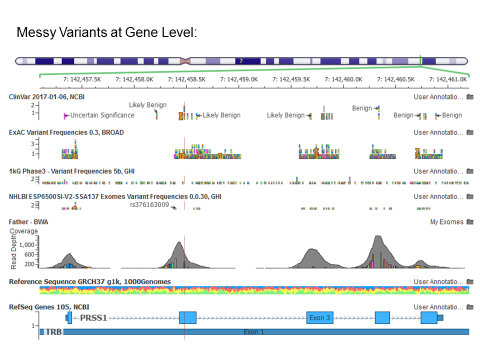
It is clear from the gene-level perspective that the variant highlighted I was investigating was not the only candidate mutation in what is documented as a highly conserved dominant-model gene.
I will skip the investigative journey and sleuthing steps that followed, but here is the upshot of what is going on:
- The 1000 Genomes consortium added a “decoy” sequence to their GRCh37 human reference that was generated by Heng Li in 2011. The decoy had real human sequence that was not properly placed in the known-to-be incomplete GRCh37 human reference. The idea was by having real human sequence align to the decoy, they would not incorrectly get aligned to second-best places along the reference genome.
- The ESP6500 exome project used this “b37+decoy” (aka hs37d5), as did the 1000 Genomes project on all their releases
- The ExAC project did not use this decoy in their alignments
- PRSS1 has a pseudogene paralog that was not fully represented in the primary sequence of GRCh37, but was captured in the decoy
- My exome and ExAC thus called many variants in PRSS1 that are not truly there, but are the best attempt to align reads for a gene that was not fully present in GRCh37
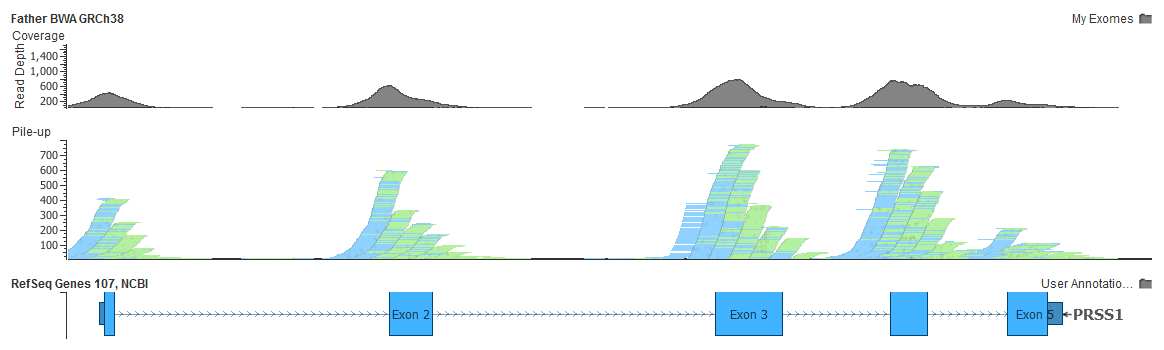
- GRCh38 no longer needs this decoy, and alignments to PRSS1 don’t suffer from these misplaced reads
As a double-check, here is the same PRSS1 gene in my exome aligned to GRCh38:
And what about ExAC’s QC flags?
This variant is flagged as InbreedingCoeff_Filter both in the online ExAC Browser and in the Filter field of the ExAC annotation source in VarSeq.
I suggest relying on this field to screen your ExAC annotations for false-positives and to potentially and carefully consider the validity of the variant calls in your own data, as I did here!
And to pre-empt questions gnomAD (ExAC expanded to 123K exomes), yes this variant shows up in the gnomAD Browser and also has QC flags applied to it as well.
We are working on curating the very recently released gnomAD exome and genome allele frequency data and will follow up with more on that shortly.
Hey Gabe,
Nice post, and you hit the key points well. This is a variant that comes up fairly regularly (I think we’ve had 4 independent emails to the ExAC help email asking “why is this variant common in ExAC?” despite the big red filter warning at the top of the variant page) so it’s good to clarify it for people.
The one thing I’d take issue with is the statement “you should consider any variant in your data that matches a QC flagged variant as if it was not in the catalog at all”. I’d rather say something like: “variants that fail FILTER status in ExAC should be treated with great caution, and in most cases should be treated as though they were not real. However, users should also be aware that variant filtering is a probabilistic process; some of the filtered variants flagged by ExAC are actually real, and some of the PASS variants are false positives”. I find that many users reify the ExAC filters as though they are ground truth – in fact those of us who spend a lot of time dumpster-diving through variants at the low end of the quality spectrum know that variant filtering remains far from perfect. The PASS/fail flags draw an arbitrary line through a continuous and noisy spectrum of quality, and there are errors on both sides of that line.
Glad to hear that you’re digging into the gnomAD data set, and we’d be very interested to get your feedback. Users of gnomAD should be aware that we’ve filtered this set using a very different analytical strategy, and overall more stringently than ExAC. Depending on user feedback we may end up rolling back the filters to something more closely resembling the ExAC permissiveness level, but we’re still debating that.
Hey Dan,
Thanks for stopping by and posting!
Yes, I totally agree with your phrasing and added a note next to mine to refer readers to it.
I really like that gnomAD has allele specific filter fields, and we used that heavily in our curation as we annotate only the alleles present in our users variants and now can provide the appropriate flags for the alternates present (in the case of multi-allelic sites).
Ultimately these types of categorical lines drawn in the continuous sand hills that we deal with at every level (same goes for functional damaging predictions) are inevitable. If we don’t do our best to pick the thresholds used to draw the line, those thresholds will be picked by users and most likely in a less informed manner.
I appreciate you and your teams dedication to improve the quality and utility in an ongoing fashion. Nothing complex is ever perfect, but things that can iteratively improve have a chance to make a lasting impact.
Hi there.
I came across your blog post when searching for how other groups are handling the variant quality flags in the GnomAD database. I am a PhD student in Australia, and my project involves trying to discover novel cancer predisposition genes in WES data from a cohort of BRCA-negative familial ovarian cancer patients, using the GnomAD database as my control reference population.
Considering what Daniel MacArthur and yourself have written above, my question is this: what do you do in those instances where you have higher confidence in the variant calls from your own exome data compared to GnomAD’s variant calls, where they might be flagged as problematic or low quality/confidence (e.g. failed Random Forest filter)? The reason I ask is that I have identified several instances in our cohort where a gene is enriched for deleterious variants within a particular gene (which I have confirmed as real using Sanger sequencing that includes flanking intronic sequences), but GnomAD has flagged a large proportion of its own variants within that gene as low quality/confidence for whatever reason. Thus I am faced with a critical issue: when comparing my variants against those in GnomAD, should I ignore all of those variants in my sequencing data that GnomAD has flagged as problematic, even though our variant calling might be more reliable (and perhaps in these instances, GnomAD might even be wrong i.e. they are real)?
Many thanks.
Hi there,
It is very possible that you have confirmed that NGS variants can be called accurately in a tricky region and if you believe your NGS target capture is similar to gnomAD then most likely the variants are being called correctly in gnomAD as well.
In other words, the QC flags are heuristic based, and as you have studied and sanger-confirmed NGS variants in a region, you may have evidence to ignore those QC flags as false positives.
One thing that would likely help is using the IGV viewer plots at the bottom of the variant details page in the gnomAD browser to look at a few of the samples containing variants and see if the alignment for those variants looks reasonable and similar to your sanger-confirmed NGS variants. That is what those “local alignment” views are for!
Gabe