An under-appreciated area of complexity when looking into the field of genetics from the outside can be found in genes and transcripts. Alternative splicing allows eukaryotic species to have a wonderfully powerful genetic code, resulting in multiple protein isoforms being encoded in a single section of DNA. But when it comes to variant interpretation, different transcripts can result in widely different predicted impacts. Additionally, interpreting a variant’s interaction with a gene also requires up-to-date annotations about gene. This includes things like the HUGO gene name (which gets regular updates) the expected mode of inheritance in the context of Mendelian disorders. We just recently updated our gene annotations and these additional gene preferences in the latest VarSeq. We also added some new features and updated capabilities to reduce the inherit complexity in working with genes and getting the correct clinical interpretation for variants.
The RefSeq Transcript Library and Reference Genome Mappings
The NCBI transcript library closely tracks every unique mature RNA (mRNA) sequence that encodes a known protein. It also catalogs many other RNA products such as non-coding RNAs that we won’t get into in this post but are included in our tracks. As a concrete example, NM_007294.4 is the RefSeq identifier for one of the five transcripts for BRCA1. It’s GenBank resource page lists the 7088bp RNA sequence as well as references to origins and related protein identifiers. But note that its definition is purely an RNA coding sequence and does not include the chromosome coordinates of the exons that are spliced into this mature RNA sequence. To use this transcript model for variant analysis, we actually need an “alignment” of the RefSeq transcript sequence to the human genome reference. In an ideal world that would be the end of it, but there are edge cases that cause problems. The two largest of these are (rare) differences between the reference genome sequence and the transcript (many of which were fixed in GRCh38), and non-unique placement of RNA sequences to the reference. See this previous blog post for more details on these issues.
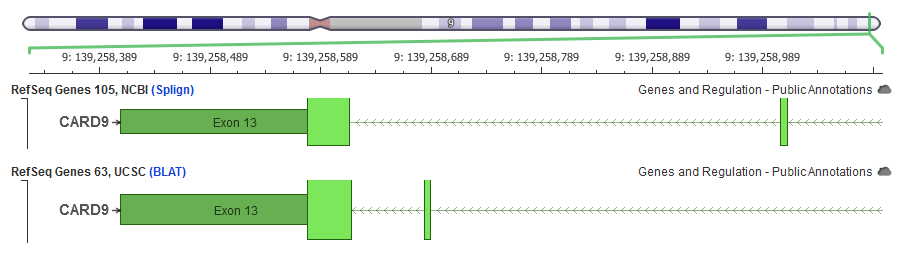
In this example, the same RefSeq transcript coding sequence has the exon 12 placed in different genomic locations based on the strategy used to align the transcript to the reference genome. VarSeq uses the NCBI alignments for its RefSeq gene tracks.
There are two human reference genomes commonly in use, GRCh37 and GRCh38. The newer human reference made an effort to match known transcript sequences when the previous reference was in error, so many, but not all of these edge cases are improved. For consistency, and for superior handling of alignment edge-cases, we use the RefSeq alignments provided by the NCBI genomes annotations team. The most recent of these for humans is Annotation Release 109 released in February 2018. These versioned annotation releases are not often updated. The last one to support GRCh37 was Release 105. Since then, the NCBI team has provided “Interim” releases that bring in updated transcript definitions. We are about to release the most recent of these that will have the version names:
- RefSeq Genes 105.20190906, NCBI (GRCh37)
- RefSeq Genes 109.20200522, NCBI (GRCh38)
Selecting the Clinically Relevant Transcript
When a gene does have multiple transcripts, there are a number of strategies for dealing with it in a variant interpretation workflow. A simple approach that works well for filtering conservatively is to always consider the highest variant impact across all transcripts. We have always supported this with our “Sequence Ontology (Combined)” and “Effect (Combined)” fields in VarSeq. But when reporting a variant in a clinical report context, it must be reported with the protein change and HGVS nomenclature for a specific transcript. Sometimes these reported transcripts are established by precedent, but other times there is little clinical data to go on. So how do we pick the default “Clinically Relevant” transcript?
Historically, VarSeq has tried to follow the heuristic used by ClinVar. We outline this in detail in this previous blog post, but one key attribute of this heuristic is whether the transcript was given an Locus Reference Genome (LRG) sequence identifier. More recently with VSClinical, we created a very information rich transcript selection dialog that pulls in data from ClinVar to get a sense of how often a transcript has been used by submitting labs to describe a variant.
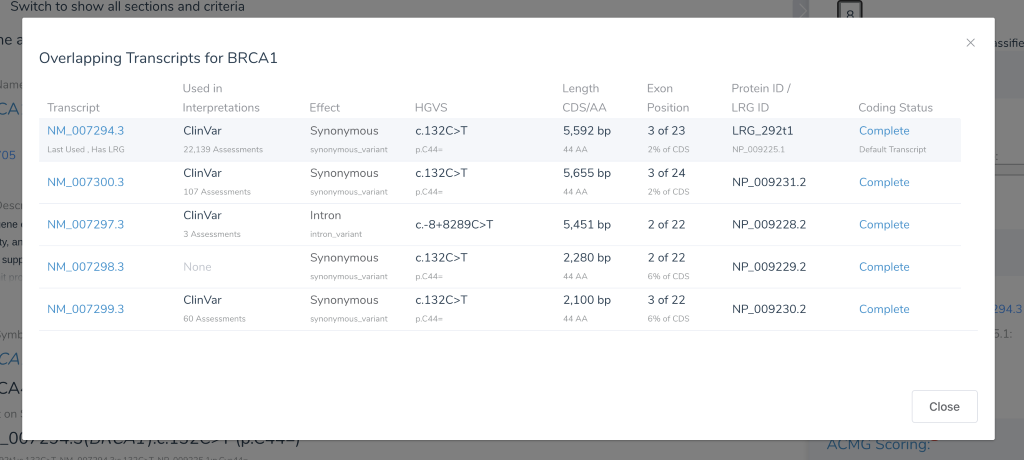
The NM_007294.3 transcript of BRCA1 has an LRG identifier and also 22,139 ClinVar assessments that describe variants using this transcript, making it a clear choice.
With the VSClinical release, we override the choice of default transcript to use any user-saved preferences (in a shared Gene Preferences file) as well as any shipped system Gene Preferences. To create our system preferences, we set the default transcript for a number of prominent genes to match the ClinVar clear majority when the existing heuristic did not.
With the release of VarSeq 2.2.1, we have incorporated a fantastic effort to improve the status quo of transcript selection. The Clinically Relevant transcripts selection heuristic has been augmented to prefer transcripts designed as “Select” by the Matched Annotation from NCBI and EMBL-EBI (MANE) project. The Select transcripts are guaranteed to be a single, high-quality transcript that is “well-supported by experimental data and represents the biology of the gene.”
In practice, there will be many genes that will change their Clinically Relevant transcript selection to now use the Select transcript from MANE. In some cases, this can result in a very different predicted gene impact.
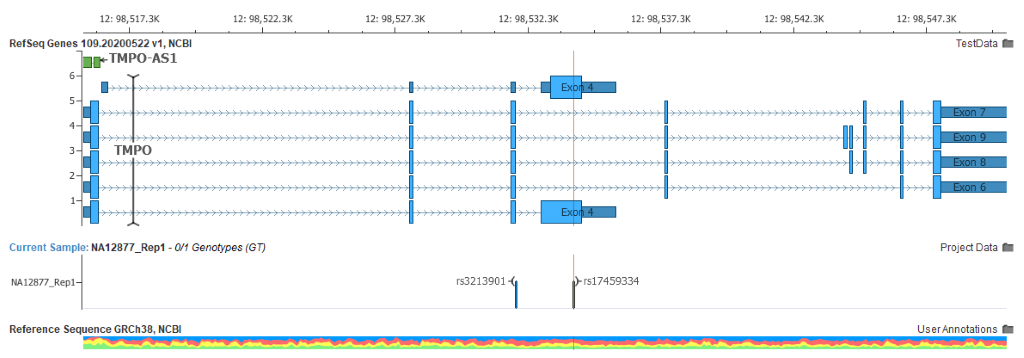
The bottom transcript was the previous default, in which this selected variant was a missense change. The MANE Select transcript is the larger 9-exon one with no exon at this position, making this variant intronic. Expression profiles and LoF variants viewed in gnomAD for TMPO corroborate that this is the correct designation.
In the upcoming RefSeq track versions mentioned above, the integrated MANE transcript set will be updated from version 0.8 to 0.9 as well.
Calling a Gene by Its Name is Harder than it Sounds
The HUGO Gene Nomenclature Committee (HGNC) defines and updates the names and symbols we use for genes. And these gene names get updated at a surprising frequency! Internally, we use more stable identifiers such and the Entrez Gene ID to track preferences for genes or to even look up annotations about genes. A gene you see referenced in an gene track as MUT may in fact now be renamed to MMUT. This in fact was an issue with the RefSeq transcript definitions released for GRCh37, as the gene symbols were a number of years out of date. Gene panels are often defined as a list of HUGO gene symbols, and VarSeq supports filtering to variants that are annotated against genes in these panels. But if these panels use a newer gene symbol, or even an older one, we risk missing a relevant gene!
To resolve both of these issues, we now update the gene names for our gene tracks from the latest information available directly from NCBI’s database. We also added a new “Aliases” field containing all of the previous gene names and aliases for a given gene and refer to that when running our “Match Gene List” algorithm when a gene symbol is not found matching the current primary gene name.
Note: We had some reported issues with the first version of the tracks with the Aliases field caused by these fields sometimes containing the primary gene name of different genes. This caused erroneous matching the wrong gene. We have since deployed new RefSeq Genes 105 Interim v3.1 and RefSeq Genes 109 Interim v2.1 that resolves this by removing any names of other genes from the Aliases list.
Gene Update Cycle
Now that gene tracks are capturing not only the latest NCBI provided transcript definitions and mappings but also the most current gene name, LRG IDs and MANE Select transcript status, we expect that we will be updating these tracks at regular intervals (on approximately a bi-yearly schedule). As always, you can choose when you update any track in your VarSeq project, and gene tracks are no different. You will receive update notifications when there are new gene tracks, but you can plan to make the update when it fits your clinical pipeline validation schedule. I’d like to thank all the feedback and suggestions from our many clinical lab customers that write in with fresh edge cases, suggestions and requests that have helped us create the tuned and ready gene tracks that we provide with VarSeq and VSClinical.



