There are many good reasons why the pursuit of the highest quality genomic interpretation would lead you to the latest human reference. It is more complete and fixes incorrect or partially missing genes that have known implications for human disease.
While most major projects cataloging human populations have plans to re-do all their genomic alignments to the new human reference genome, the reality is that if you want to use GRCh38, there are currently no existing population catalogs with variants called natively in your coordinate space.
Some critical annotations are on GRCh38, especially those sourced from the NCBI. These include ClinVar, RefSeq Genes, and dbSNP. But population frequencies remain a very large gap. To fill in the gap, we at Golden Helix have for many months been working through the minute details of getting very accurate and allele count corrected population catalogs and other critical interpretation resources lifted over and made available for the new human reference.
Fixing Variants and the Reference is Minor Case
In the recent update talk at ASHG, Dan MacArthur, the lead of the gnomAD/ExAC team, said that they are working on a future release of 65K genomes mapped and called natively on GRCh38.
https://twitter.com/saumyads02/status/921129262425526272
Similarly, there has been for a long time some work to re-align the 1000 genomes raw sequence data to GRCh38. Yet at the end of the day, these projects are ongoing and there are currently no GRCh38 coordinate variant population frequencies available from 1000 Genomes, ExAC or gnomAD.
We mentioned earlier that we have implemented our own version of the UCSC LiftOver algorithm in VarSeq. This feature is available in our convert wizard in the final step as an Advanced option:
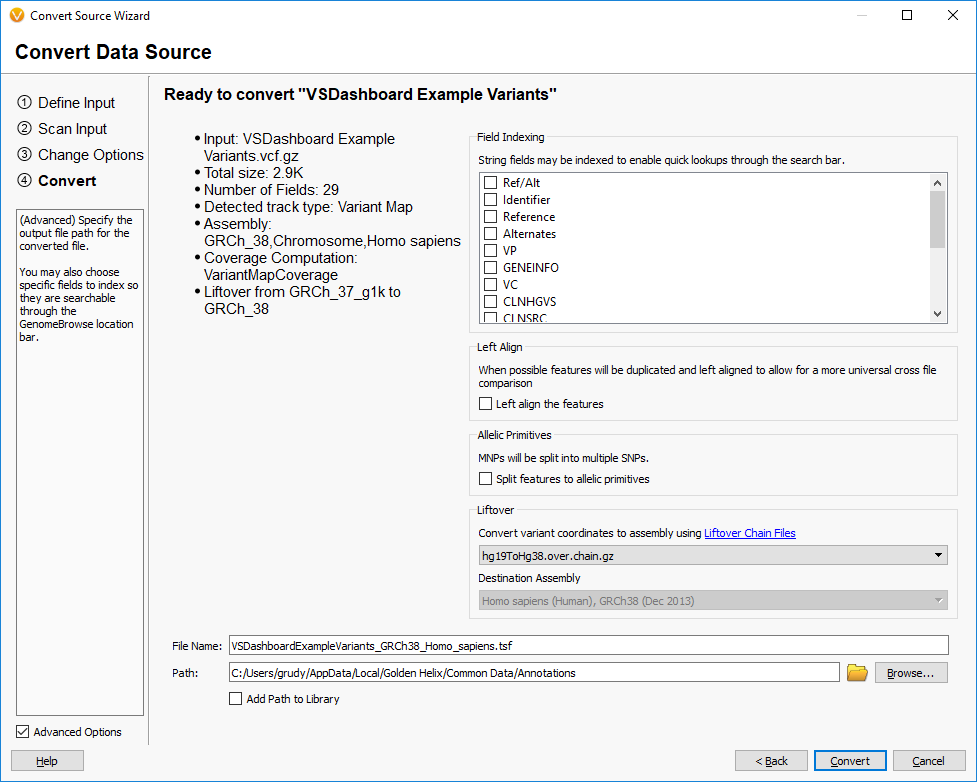
This feature improves on the base LiftOver algorithm by properly handling the mapping of both the start and stop of features to the new coordinate system and also being aware of the possibility of alleles being flipped in the reference for variants.
Why does this matter? There were a number of changes to the GRCh38 reference where an allele that turned out to be the “minor” allele was part of the human reference. This is inevitable when creating the reference from individual humans, who will naturally sometimes harbor rare or even “private” (not seen in anybody else) mutations.
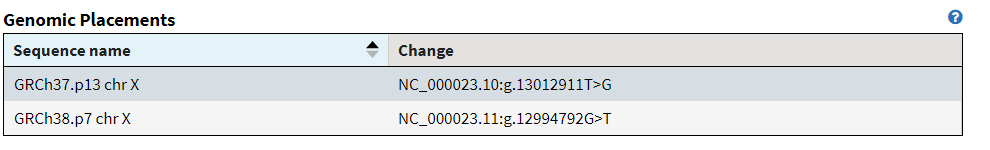
An example of this is rs5934051, which not only used to have a T at position 13,012,911 on chromosome X, but that T never occurred in anybody else as everyone actually had a G. In fact, in the corrected allele frequency tables for 1000 genomes and gnomAD, these “variant” (now a G>T) occurs with an allele count of 0.
In many other cases, some people actually had the reference allele, but the vast majority of folks had the “alternate” and in the new reference sequence, that alternate allele is the new reference. In these cases, we go from the alternate allele frequency being greater than 50% (and not being the minor allele) to the alternate allele frequency being less than 50% (and now being the minor allele).
Clearly when lifting over critical population frequency data, we want to have our annotation counts and frequencies reflect the reference alleles in these cases, and so we have been carefully writing the bioinformatic transformations to do just that.
The Wait is Over
The end result is that from VarSeq, you will now be able to annotate against the population catalogs you have come to rely on while still using the GRCh38 reference and trusting that the frequency and allele count information is accurate!
Note that this goes beyond just flipping frequencies (0.99 -> 0.01), but also includes updating the allele count, heterozygous count and homozygous count for the entire catalog and each sub-population. Recently I noted that we improved upon the high-level frequencies counts default available for the 1000 genomes catalog and included the genotype counts for each sub-population. Those counts are also available in our GRCh38 version of the track and correctly adjusted for flipped alleles.
You can expect to find the following tracks in our repository for GRCh38:
- Population:
- 1000 Genomes (Phase 3 with our Genotype Counts)
- NHLBI ESP6500
- ExAC 0.3
- gnomAD Exomes and Genomes
- dbSNP
- Clinical:
- RefSeq Genes
- ClinVar
- GWAS Catalog
- OMIM Variants and Genes
- Functional Predictions:
- dbNSFP Functional Predictions 3.0
- dbscSNV splice altering predictions
Finally, here is another example of the above population catalogs on GRCh37 for an allele that was clearly a “reference in minor” situation (labels are set to the alt allele frequency):

And here the same rs2109135 variant in GRCh38 with all of our allele flipped and frequency corrected tracks:

I look forward to supporting the new and improved human reference as labs consider the migration of their bioinformatic pipelines and workflows to be natively based on GRCh38!




Dear,
I have a question .
I have an exome data set aligned with Hg38, and I annotated with Annovar gnomAD 38 without problems , but I have many variants with “problems” when I used Hg38 gnomAD Version 2. I think that my problem and explication could be that gnomad version 2 was constructed using Hg37 and versions of Hg38 of gnomADv2 are a lift over of gnomADv2 constructed with Hg37. Do you have experience on this ?
Thank you.
Hello Pablo,
If you are still having problems with the build, feel free to reach out to [email protected] and we are happy to take a look at your workflows!