Variant Normalization: Underappreciated Critical Infrastructure
It may surprise you to learn that every variant in the human genome has an infinite number of representations!
Of course, although true, I’m being a bit hyperbolic to prove a point.
Even seemingly simple mutations like single letter substitutions are legitimately represented differently in the local context of other mutations that can be described as a single complex substitution, or alternatively as multiple single letter changes mixed with insertions or deletions or even potentially equally represented along different sections of a repetitive reference sequence!
While some simple rules and heuristics adopted across bioinformatics tools help, the rough reality is that whenever you are merging variants from multiple variant callers, and in some cases with the same variant caller but from different samples, the disharmonious representations can obscure the pertinent facts about which variants are shared between samples and at what frequency they occur in your cohort. Variant normalization is therefore foundational to accurate NGS data analysis.
As we have been developing VSWarehouse as a genomic warehouse — a single variant store to accumulate variants from all your relevant clinical and research samples, we have invested time in the core VarSeq variant normalization, merging and collapsing transformations.
Our latest release of VSWarehouse and VarSeq now contains these improvements, and I’d like to share through example some of these edge cases and how they can show up in your data.
Getting it All Right
When merging multiple VCF files with multiple samples together, whether initially on importing data or when adding new samples to an existing warehouse of samples, we have the following goals:
- Variants are represented in their “simplest” form, both so they can match up with the annotations of other populations and variant databases, and also so they can be used as a population frequency annotation source itself
- Variants should preferably contain one alternate allele, but when genotypes exist with multiple non-reference alleles, this is captured with accurate genotypes and allele frequencies
- The same variants are always merged and represented in a single canonical form so sample genotypes and allele frequencies are accurate
- All transformations preserve supporting allele-specific data such as per-allele read counts, quality scores, derived variant frequencies, etc.
To achieve these goals, we actually perform a pipeline of independent but mutually supporting steps when importing and merging variant sources:
- Merge samples from multiple sources
- Break complex variants down to “allelic primitives”
- Split multi-allelic variants in a sample-aware manner using strategies appropriate for cancer (somatic) vs hereditary (germline) variant workflows
- Left-align variants into a canonical position
- Collapse variants that are the same after normalization, merging sample data
There are multiple tools that have tackled this problem and contain some of these components, but I believe VarSeq is the only one that puts them all together with a focus on the utility of the sample level data for end-to-end variant analysis.
The popular GATK suite has a tool called LeftAlignIndels that does, well, what it says it does!
The tool “vt” is described in Unified Representation of Genetic Variants and does left aligning (although not with a Smith-Waterman local re-alignment like VarSeq does) and reducing to allelic primitives.
The suite of tools in vcflib related to variant representation is probably the most comprehensive of the open source bunch. It does a SW left-align (that’s where we got the idea!), allelic primitives and multi allelic splitting.
Our own implementation of these algorithms was largely inspired by vcflib, and expanded to handle the many edge cases of different variant callers and to preserving allele and sample-specific data through all of these transforms.
Finally, we found the need to add a variant collapsing step, as complex variants from different samples and different callers may result in multiple normalized records for a single variant with the sample data split between the two.
Pictures Worth a Few Thousand Lines of Code
I have previously written about the Supercentenarian cohort of 17 whole genome sequenced individuals over 110 years of age and even a follow up where we tackled some of the details of allelic primitives.
This is the dataset that keeps giving, as the combination of breaking complex variants down to primitives and left-aligning them sometimes resulted in two variant records that need to be merged with our new Variant Collapsing transform.
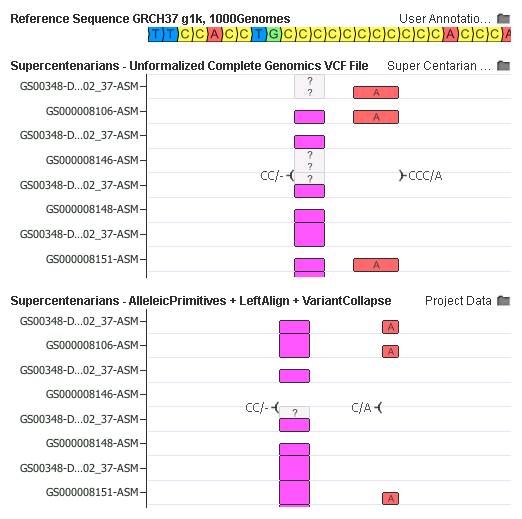
In this example, the complex CCC/A mutation was broken down into a CC/- and C/A mutation. Once left-aligned, the CC/- is collapsed in a phased-genotype aware mode at the sample-by-sample level.
What’s amazing about this example is how the interpretation may have been incorrect before the normalization. Notice how the CC/- deletion were transformed from a heterozygous to homozygous state for samples GS000008106-ASM and GS000008151-ASM as the complex variant nearby was broken down merged (The phased 0|1 and 1|0 genotypes were merged to a 1|1).

In this example, the import of the ordering of the transforms is apparent. A multi-allelic variant TTTTTTTTT/TTTTTT/-/TTTTTTT is first broken down into three different variants, normalized, then the left-align dramatically changes their position. In the canonical form, 2 of the 3 variants have records in dbSNP and thus will receive correct and useful annotations.
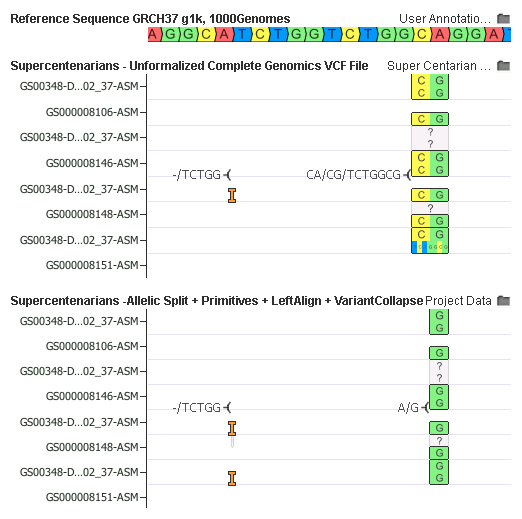
Finally, in an example that shows the work of all the transform steps, the raw VCF up top has two variants. Note the right one is a multi-allelic CA/CG/TCTGGCG and sample GS00348-DNA_F01_202_37-ASM has both alternate alleles.
After all transforms are down, we result also in only two variants, and they look surprisingly different!
Here is how it works:
- First, the multi-allelic transform split the second variant down to a CA/CG and a CA/TCTGGCG
- Then allelic primitives trimmed the CA/CG to A/G and then broke CA/TCTGGCG down to an insertion of a -/TCTGG, followed by the SNP A/G (Note there are 4 records at this point)
- Left-aligned using Smith-Waterman was cleverly able to shift the -/TCTGG insertion 10bp to the left
- And finally, the Variant Collapsing reemerged in the genotypes of the now duplicate insertion and SNP records to result in just two variants
It’s also now possible to both visualize and annotate/interpret that DNA_F01_202_37-ASM is homozygous for the common A/G SNP and heterozygous for the rare insertion!
Fundamentally Necessary, Inherently Complex
It may be easy to dismiss these examples as specific to the somewhat stand-alone way in which the CGI variant caller prefers complex variants to primitives, but I can assure you that I have seen similar examples in GATK, FreeBayes, Illumina Issac and the Ion Torrent variant callers.
Especially important is the need for a single canonical variant form when merging samples and creating your own reference population and warehouse of samples that can be used to annotate the frequency and genotype counts of known variants in the interpretation of your new samples. This is precisely the workflow VSWarehouse was built to support.
While I demonstrated success stories of complex variants being broken down and represented canonically in identical manners, it is possible to construct situations where two different variant callers did their own different allelic primitive algorithms which result in variants with irreconcilable normalized forms. The only solution here is to reconstruct the larger localized sample haplotypes and choose a representation such as a “graph genome” or even a consistent variant normalization transform before doing sample comparisons.
I look forward to seeing how the progression of this work continues in the GA4GH variant benchmark working group.
In the meantime, I am very pleased with the breadth and capability of the strategy we have matured in VarSeq and VSWarehouse that is immediately available to all our users.
Do you have an example variant where it can only be left aligned with SW but not with the algorithm in vt normalize?