Thank you to everyone who joined us for our webcast on the upcoming VarSeq features supporting the full spectrum of genomic variation! Traditionally, NGS cancer testing started with small gene panels that looked at a small set of the most common genes to identify small mutations, such as BRAF V600E. However, there are many classes of mutation that cannot be captured by these small gene panels. This shortcoming is addressed by a new generation of cancer tests called comprehensive genomic profiling tests. These tests analyze a broad panel of genes to identify the four main classes of genomic variation known to drive cancer growth:
- Small Variants
- Copy Number Variations
- Large Insertions
- Complex Rearrangements
These tests also consider genomic signatures, such as tumor mutational burden and microsatellite instability, which provide an indication of the tumor’s genetic progression.
In the upcoming VarSeq release, we will support every class of genomic variation, including genomic signatures. Small variants, CNVs, and complex rearrangements can now be imported from a single file and performance improvements have been made to support faster import of whole genome sequencing data. Additionally, you can annotate and filter all of these mutation types using VarSeq’s comprehensive annotation catalog and powerful filter chain. The set of filtered mutations can then be imported into VSClinical where they can be analyzed in accordance with the AMP Guidelines and included in a customizable clinical report.
VCF Breakend Notation
In the upcoming release, VarSeq will support the import of complex rearrangements from VCF files, which typically encode rearrangements using breakend notation. This powerful notation is capable of describing the full spectrum of structural variation. This includes the following mutation types:
- Duplications
- Deletions
- Translocations
- Inversions
- Large Insertions
In this format, a rearrangement is defined as a pair of novel adjacencies, called mates, which are tied together by their position and id. Each breakend’s ALT field specifies the alternate sequence t, along with the position, p, and the orientation of the joined sequence as follows:
- t[p[ → piece extending to the right of p is joined after t
- t]p] → reverse complemented piece extending to the left of p is joined after t
- ]p]t → piece extending to the left of p is joined before t
- [p[t → reverse complemented piece extending to the right of p is joined before t
The example below shows three complex rearrangements encoded as six breakends and demonstrates all possible breakend orientations.

The genomic interpretation of the above breakends is illustrated by the image below:
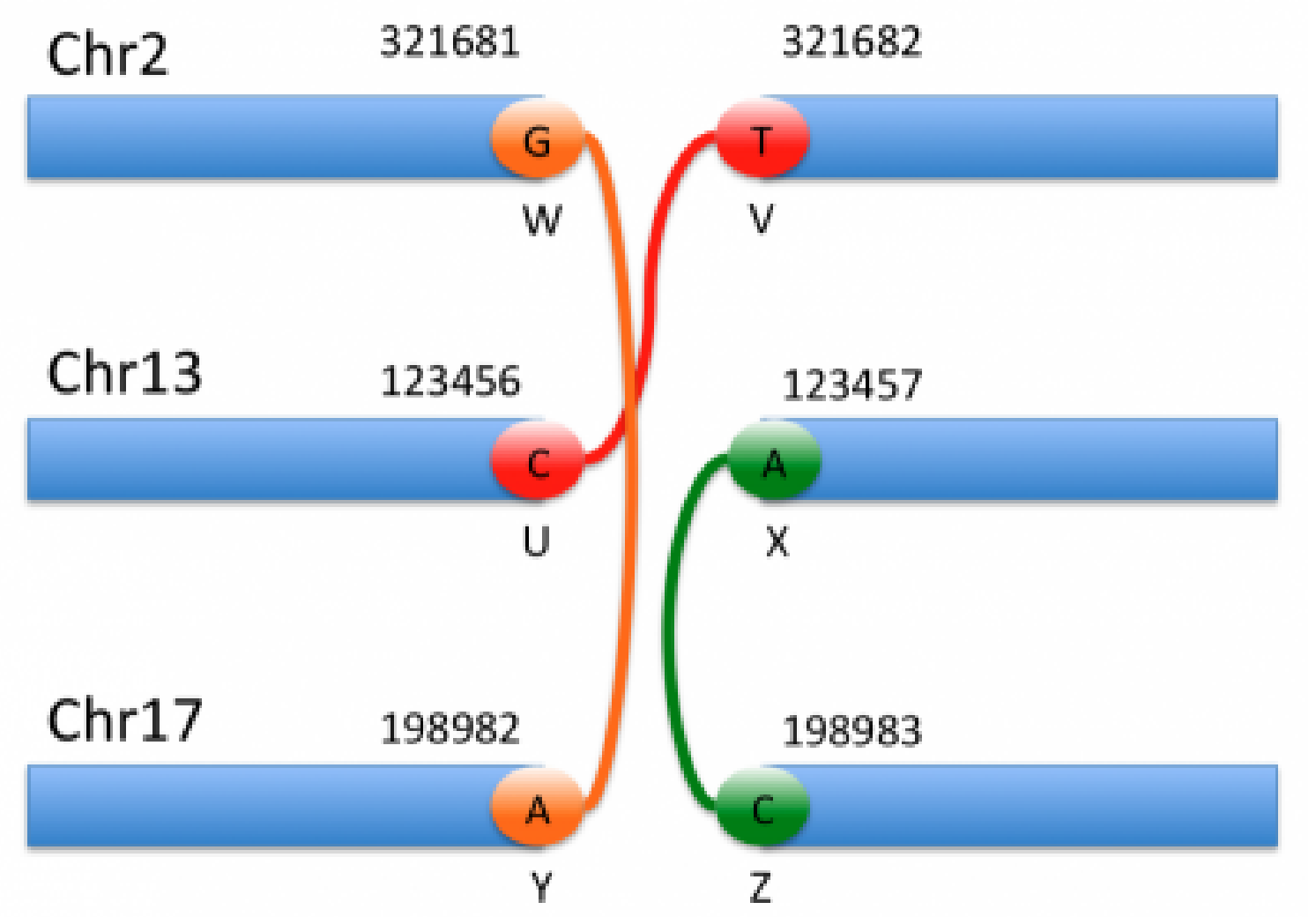
By looking at the specifics of this notation alongside the figure above, we can begin to decipher the notation and interpret each event. For instance, the event shown in orange corresponds to the 1st and 5th entries in the VCF notation. Based on the ALT field of the first entry, we can see that this is a translocation in which the mate is in chromosome 17, and the reverse complemented piece extending to the left of the mate is joined after the base G in chromosome 2.
The above example illustrates the complexity involved in reasoning about these breakends. Thankfully, VarSeq reduces this complexity by automatically joining mate pairs and inferring the type of each rearrangement on import. Additionally, the VarSeq gene annotation algorithm identifies all possible fusion gene pairs and predicts the effect of each fusion on the protein product. As noted above, CNVs can also be described using this notation. In these cases, the CNV events will be imported into both the Breakend and CNV tables so that they can be annotated and interpreted in either context.
CRAM File Support
To address the massive amounts of storage space required by whole genome sequencing, clinicians and researchers need a way to compress genomic data into smaller packets of information. The CRAM file format for storing read alignments addresses this problem. For a given read in an alignment file, the vast majority of nucleotides will exactly match the reference sequence. Thus, if we avoid storing these matching sequences, we can greatly reduce the storage size required by the alignment file. This is the primary innovation of the CRAM file format: it compresses alignment data by only storing potions of reads that differ from the reference sequence. This process is illustrated by the image below:
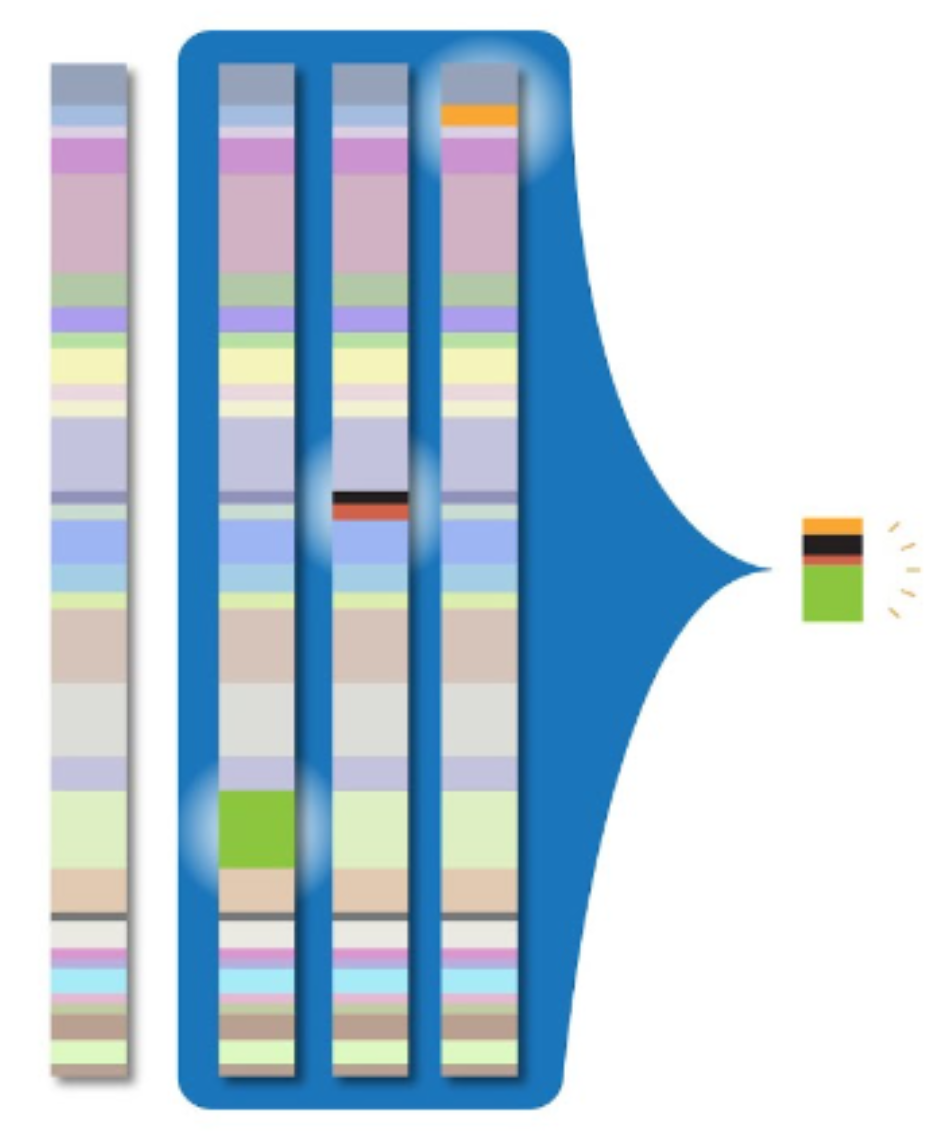
While this greatly reduces the storage size of alignment data when compared to the BAM file format, the downside to this approach is that it requires decompression, which results in slower read times.
In the upcoming release, VarSeq will fully support the CRAM file format. This means that users will be able to import and visualize CRAM files. However, this feature does require that the reference track exactly matches the reference sequence used during alignment. Many labs use custom reference sequences during alignment that have been modified to mask certain regions. CRAM files generated from reference sequences that employ this sort of custom masking will require the creation of a custom reference sequence track. That said, VarSeq will have built-in support for the most common alignment reference sequences for both GRCh37 and GRCh38.
Conclusion
As part of our upcoming VarSeq 2.3.0 release, we have greatly improved our analysis capabilities by supporting the import, annotation, and filtering of mutations across the entire spectrum of genomic variation. This includes support for concurrent importation of small variants and CNVs as well as complex rearrangements encoded using breakend notation. These improved import capabilities also include support for the import and visualization of CRAM alignment files, which provide a significant reduction in required storage space compared to the BAM file format. This release also strongly supports structural variant annotation, filtering, and interpretation, including structural variant effect prediction. Finally, all clinically relevant structural variants can be interpreted using the VSClinical AMP Guidelines workflow and included in the final clinical report.
Thank you again to all of the attendees and customers who have helped guide our development process. If you have any additional questions or follow-ups, please don’t hesitate to contact us at [email protected].
Golden Helix has developed innovative tools for the clinical interpretation of variants. VarSeq 2.3.0 will deliver powerful capabilities for genomic profiling in cancer and for simplified importing, annotating, and filtering mutations across all spectrums of genomic variations. Please click the VarSeq icon below to learn more.
