Thanks to all of you who were able to attend the live webcast introducing the newest genomic analysis tool within VarSeq, VSPGx! For those of you who could not make it to the live presentation and demonstration, I will fill you in on what we covered. If you would like to watch or re-watch the webcast, you can access it here!
Understanding Pharmacogenomics and Test Outcomes
Nathan Fortier kicked off the webcast by introducing pharmacogenomics as a discipline and how pharmacogenomic testing can be used to predict how an individual will respond to a given drug, determine appropriate dosing, and assess whether an individual is at risk of toxicity if prescribed a drug. The results of a pharmacogenomic test have three main components: a diplotype for each tested pharmacogene, the individual’s metabolism phenotype based on the diplotype, and a drug treatment strategy recommendation based on the combined phenotypes across the tested pharmacogenes.
Diplotypes and Star Alleles
Defining the named alleles present in each pharmacogene is the first step for NGS data and PGx testing. Usually these alleles are typically described using star allele notation, which identifies pharmacogenomic markers by means of a designated number for a given gene. The combination of two haplotypes (named alleles/star alleles) is the diplotype. Throughout the webcast, we reference the CYP2D6 gene as an example (Figure 1).
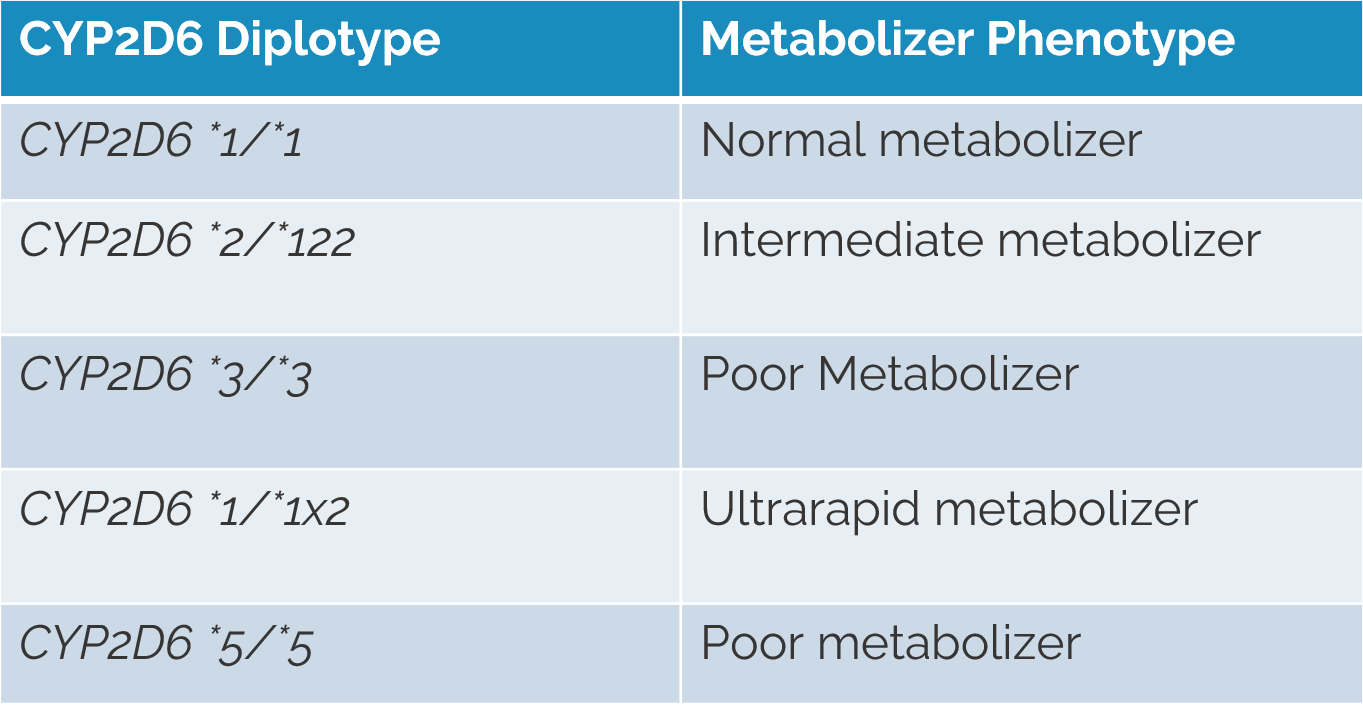
PGx Variant Detection and Recommendation Algorithm
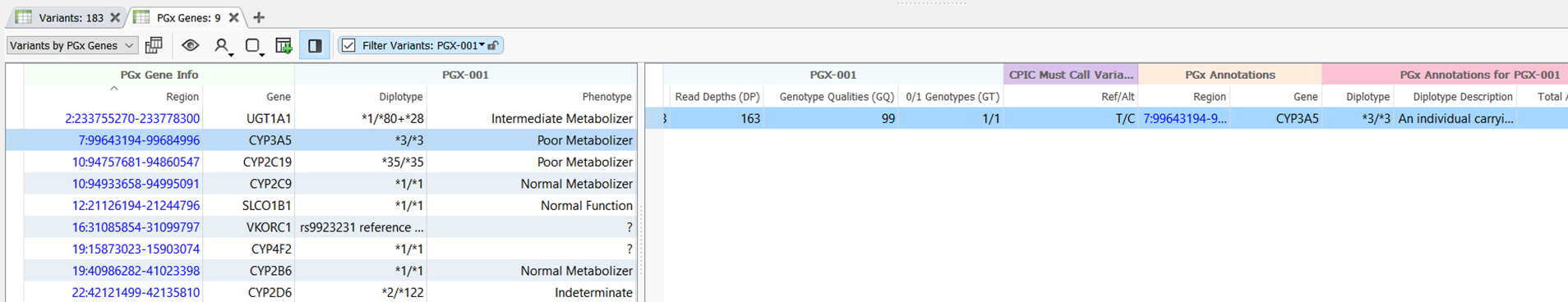
In the demonstration, we showed the all-new PGx Variant Detection and Recommendation algorithm, which identifies pharmacogenomic diplotypes and then annotates them against drug recommendations. By default, diplotypes are annotated against recommendations provided by The Clinical Pharmacogenetics Implementation Consortium (CPIC), as they are the most comprehensive source of allele definitions and recommendations. However, custom annotations can be used, provided that the required fields are present. An example of the output of the PGx Variant Detection and Recommendation algorithm is shown in Figure 2.
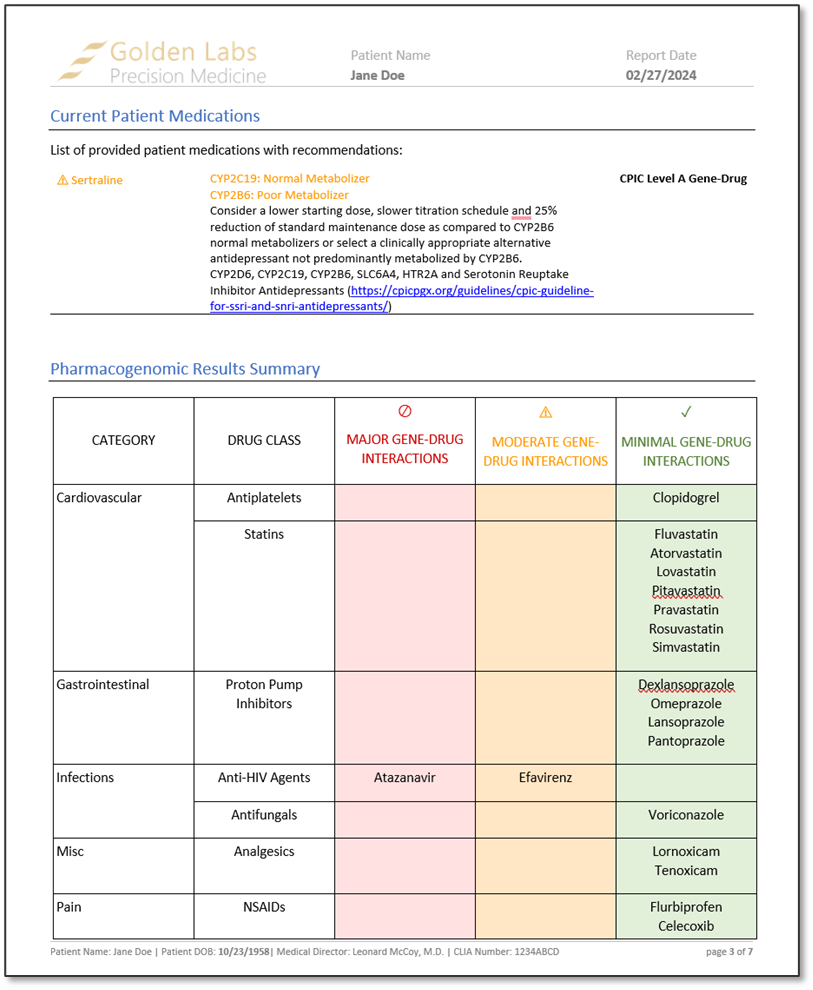
After reviewing a couple of examples of the output of the algorithm, we rendered a word-based PGx report in VarSeq. The report contained information about how to interpret the report findings, current patient medications, placement of each drug that was associated with the diplotype in a table categorized by minimal, moderate, and major gene-drug interactions, CPIC recommendations for each drug based on patient phenotype, and a list of alleles tested and not tested. These reports, much like the word-based reports, are fully customizable.
Answering Your Questions
We did have a few questions at the end of the presentation that I will go over again to make sure everything was answered!
Q1. What is the required input file for VSPGx?
A: gvcf or vcf files are the preferred inputs for VarSeq
Q2. Are there any issues handling things like the gain of function variants? For example, with CY2D6 three or more alleles?
A: CYP2D6 can be a tricky gene as there are several relevant alleles that are defined by gain of function SNPs and structural variants. While VS doesn’t currently support calling structural variants, you will be able to import them via a sample manifest.
Q3. Can VSPGx handle G6PD, which is an X-linked gene?
A: We do support handling a variety of genes, including the G6PD gene as there are CPIC recommendations and defined alleles for the G6PD gene.
Q4. What is the price for VSPGx?
A: Please visit the VSPGx product page on our website and fill out an evaluation form to request a demo with one of our area directors, and they provide more information.
Q5. Does VarSeq import data from arrays such as TaqMan OpenArray?
A: Yes. While VS does not natively have the ability to support this kind of data, we do have secondary analysis scripts that convert the raw array data into a vcf file format that can be used in VarSeq.
We are so excited to welcome the complete pharmacogenomic workflow to the VarSeq suite and hope you are too!
If you have any questions, please email our team today at [email protected].



