When a variant shows up as rare in the general “healthy” population, as indicated by low frequency or absence in one or more commonly referenced population catalogs such as GnomAD Exomes or 1000 Genomes, this indicates by proxy that the variant may be pathogenic. However, several factors determine the frequency threshold below which a variant is considered rare enough to be pathogenic when going by the ACMG guidelines. One has to take into account factors such as variable mode of inheritance based on gene-disease associations, the zygosity, the matter of penetrance, and of course, what is generally accepted in the literature or submitted evidence for that variant or gene.
These factors make assigning these criteria less objective and more variable on a case-by-case basis. The benefit of using VSClinical’s ACMG classification system is that while we do offer a large degree of automation in classifying variants, we also offer complete transparency and flexibility. As such, a user could account for the subjectivity of each case they are evaluating. This is a common topic that comes to our support inbox, and below, I discuss three ways in which a user can leverage the transparency and flexibility of the VSClinical ACMG tool.
Adjusting Frequency Thresholds
When evaluating a variant in VSClinical, you have the option to “Score All Recommended Criteria,” thereby applying our automated classification and interpretation to that variant. Several of the ACMG criteria are based on the population frequency of variants.
By default, VSClinical will be using specific thresholds based on the ACMG guidelines to determine what is a common versus a rare variant. These thresholds are different for genes inherited in a dominant versus recessive mode. In some cases, variants may have incomplete penetrance for a disease and so a higher population frequency may be tolerated for a potentially pathogenic variant. Therefore, we give the user the option to adjust these frequency thresholds for specific ACMG criteria. Moreover, users have the ability to customize the population frequency catalogs used as the basis for alternate allele frequencies.
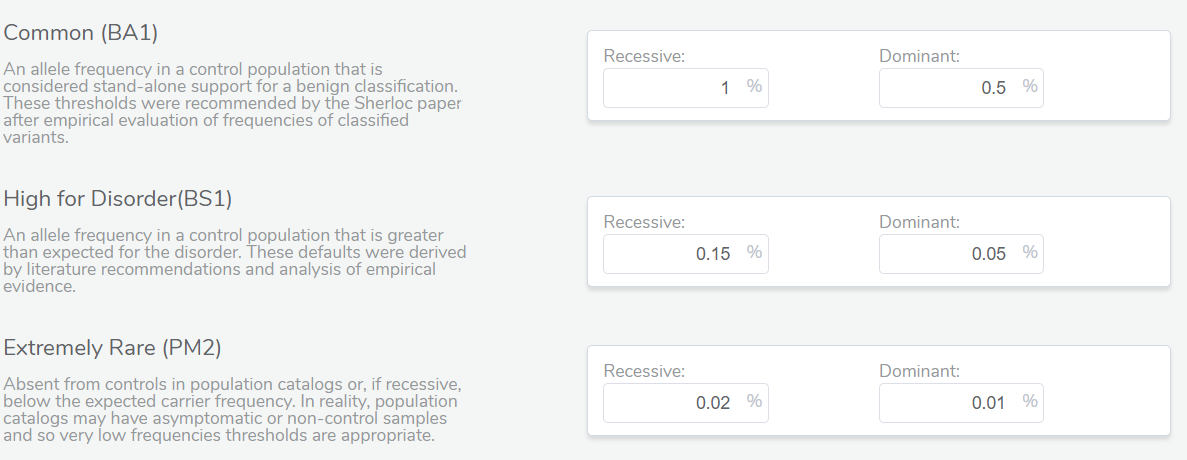
Adjusting Inheritance Mode
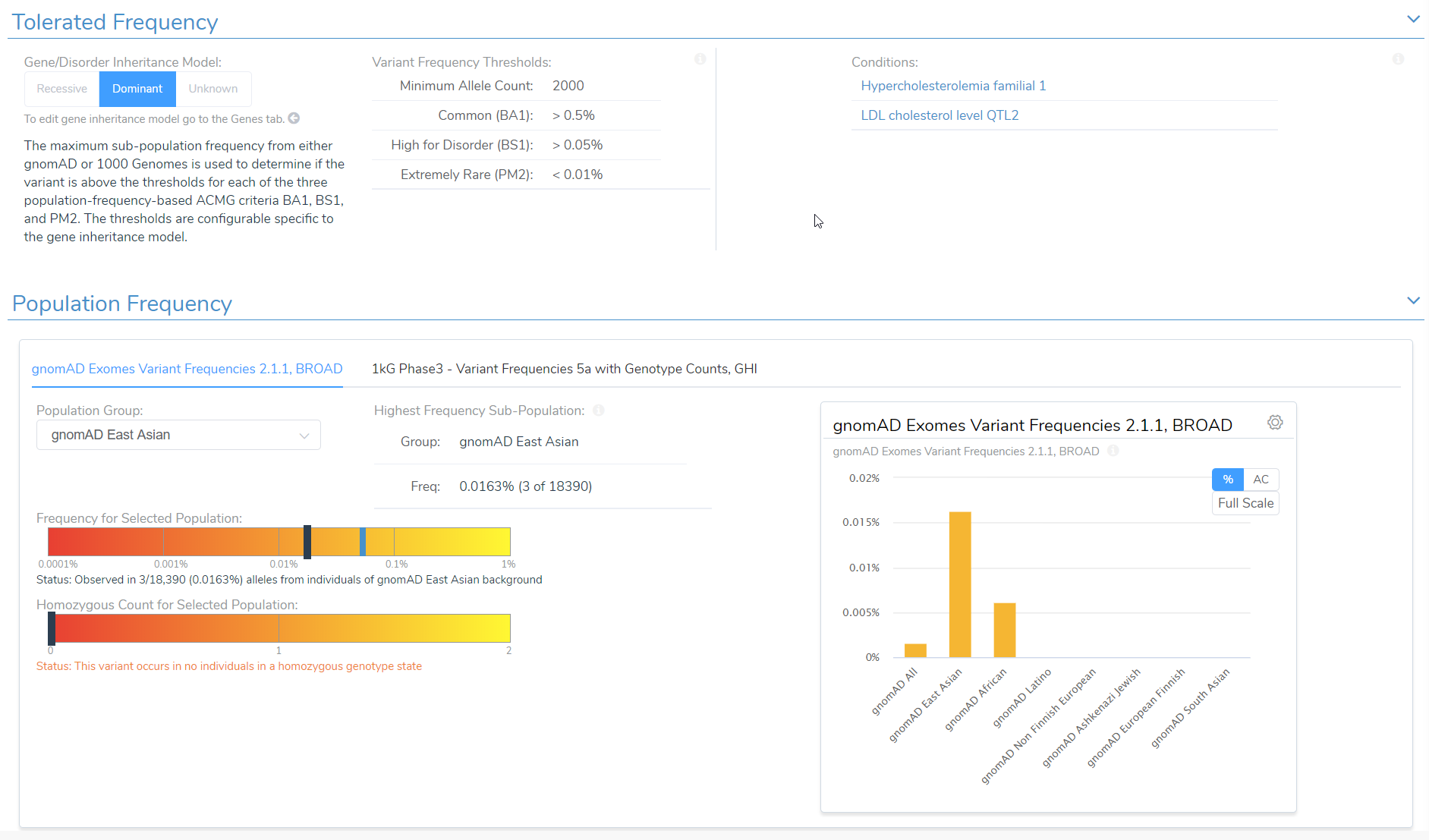
In the example below, we have a splice variant in the gene LDLR with an allele frequency of 0.016%, which happens to be homozygous in the sample. Interestingly, no homozygous individual had been reported before in the population catalogs used. Based on the most prevalent disorders associated with this gene, the inheritance model is dominant, and to be considered rare, the variant must have a population frequency of less than 0.01%. As such, the variant barely missed the cutoff. However, with a recessive model, the variant would be considered rare. In cases like these, the user’s discretion and what is known about the disorder being studied in that particular case would factor into the final classification. This is where the transparency and flexibility of VSClinical are apparent, as we allow the user to adjust the inheritance mode. Adjusting this variant to a recessive inheritance model would cause it be assigned the PM2 criteria for being absent or at very low frequency in the population.
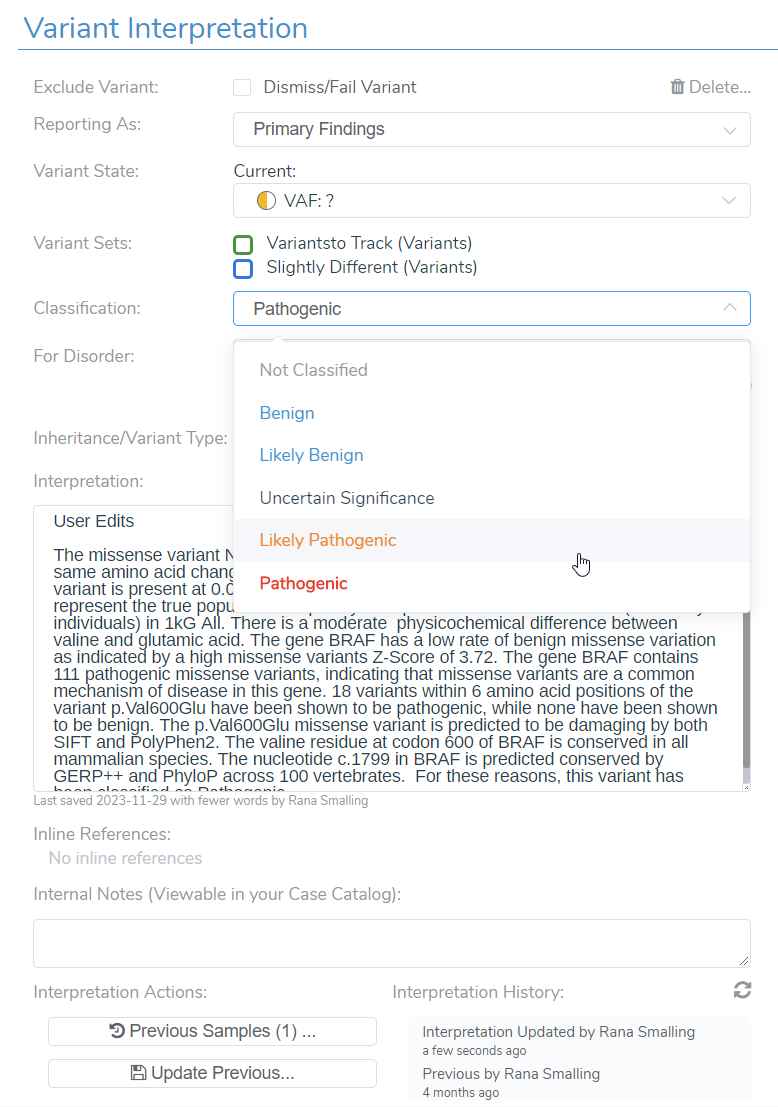
Manual Selection of Criteria and Final Classification
Another way a user may leverage the transparency and flexibility of VSClinical is by using the manual override as well as the wealth of evidence brought into the software, such as ClinVar evidence and literature references in VSClinical. VSClinical allows the user to add additional criteria, such as PS3, based on any in-house laboratory studies or new evidence. They are also free to manually upgrade or downgrade the recommended classification. VSClinical provides enough evidence and transparency to allow a user to have confidence in their reporting. A user is also allowed to save notes to a variant interpretation, especially if they have made changes deviating from the VSClinical automated classification and interpretation. The changes and notes will all be saved to your interpretation or variant assessment catalog for future use and for sharing with colleagues.
For any questions about this topic, please reach out to [email protected]. Thanks for reading!



